Acknowledgments
The great increase in computational chemistry used in the classrooms of the world has been due to thousands of professors, teachers, assistants, students and others influenced by academia. Many of these people go unthanked throughout their careers. This primer is dedicated to all of those people concerned with the education of students at any level.
Middlebury College has provided me with opportunity to contribute to the curriculum of organic chemistry. For this I thank the school and all involved. I would like to thank, more specifically, the following people for their support, understanding and enthusiasm towards education:
Janet Nelson for aiding my exploration and education in both Molecular Modeling and the field of teaching.
Jeffrey Byers for the interest in expanding computational chemistry to the realm of organic chemistry both at the introductory level as well as the research level of radical chemistry.
Steven Sontum for understanding the computational and mathematical aspects of Molecular Modeling and supporting my interest in providing this primer for organic chemistry.
The seniors and Feb. class of 1996.5 for all the entertainment.
And my parents, my sister and Christina for all their support, encouragement and understanding.
Financial Support from: NIH, VISMT-ILI, Middlebury College Senior Work Fund
Introduction
The growth of molecular modeling at the undergraduate level has been tremendous over the past twenty years. The more recent advances in molecular modeling are of particular importance. The current computer programs allow students to visualize calculated molecules as they might appear in a reaction, or as structures bound in calculated bond lengths, angles, and dihedral angles. The ease of use, the speed, and the computational power available has allowed academic institutions, like Middlebury College, to implement molecular modeling laboratories.
Numerous articles involving molecular modeling have appeared in journals ranging from Chemistry and Engineering News(1) to The Journal of the American Chemical Society.(2) The topics of these publications range from undergraduate chemistry questions for teachers to pose to students(3) to molecular structure determination.(4) Educators are beginning to use molecular modeling as an pedagogical tool. The ability of many programs to teach students about the properties and subtleties of molecules draws professors to this field of chemistry. A partial list of possible properties includes not only molecular structure, but molecular orbitals, including Highest Occupied Molecular Orbitals (HOMOs) and Lowest Occupied Molecular Orbitals (LUMOs), molecular symmetry, bond lengths, bond angles, bond dihedral angles, estimations of the heats of formation, electronic structure, and numerous other chemical traits.
The use of molecular modeling in educational facilities is gaining momentum. When E. M. Engler, J. D. Andose and P. v. R. Schleyer published the their paper "Critical Evaluation of Molecular Mechanics",(5) Computational Chemistry began its current journey into the classroom. In 1994, at the University of Abertay Dundee, UK, Dr. N. Ringan determined "Computer-Aided Molecular Design (CADM) ... is a useful addition to (the) final-year B.Sc. honours courses in chemistry and biochemistry."(6) The CADM was needed "to insure that graduates are exposed to the concepts and terminology of this discipline."(6) Honors students of Chemistry and Biochemistry use Computational Chemistry because it is so common in the fields of research and development. The goal of the class is to "provide an overview of Computational Chemistry."(6) Not only are curricular developments changing in the undergraduate course of study, but the software and hardware are available at reasonable prices to students and their colleges.
At California State University in Los Angeles, Dr. Casanova has his introductory organic class examine fifteen different pre-calculated molecules. These simple viewing exercises give the students the opportunity to "see" molecules and examine bond lengths, angles and dihedral angles.(7) Professor Box of City College of C.U.N.Y. states that Computer-Assisted Molecular Modeling (CAMM) studies are done before experiments or synthesis of complex materials with greater frequency than before.(8) Box supports Casanova's approach to modeling with ranges of molecules. He feels that the CAMM exercises should not only use the available computer technology, but require the experimenters to use structures from X-ray crystallography studies to note chemical bond lengths, angles, natural shape and variations between the molecules. In a combined study, molecular modeling and X-ray crystallography, the students "can then approach structural organic chemistry with a deeper understanding of the dynamic interplay of stereoelectric effects and structure."(8)
The organic chemistry curriculum is not the only undergraduate course being augmented by this technological advance. Biology and biochemistry classes use SPARTAN's amino acid library to investigate these important biological molecules. Inorganic chemistry syllabi have contained investigations using the resources of the Expert Builder and PM3(tm) method of Semiempirical Calculations.(9)
An example of the breadth that molecular modeling within education reach is displayed in T. W. Hanks's paper Models of 2-Butanone: Using Graphics to Illustrate Complementary Approaches to Molecular Structure and Reactivity.(3) This paper explicitly displays that the possible uses of an orbital view of molecules aids the students in organic chemistry. Extensive studies of one molecule, 2-butanone, were done spending only computer time, not laboratory time on synthesis or analysis of the compound. A process that may involve multi-step synthetic techniques in the lab and take hours to complete, is finished in less time by building structures through the computer and submitting relatively short calculations. The preparation and reactivity of molecules can, and should, be later determined in the lab, but the many first steps, as suggested by Box, Cassanova and Hanks, are now taken with molecular modeling packages.
Improvements in the quality, speed and output of the experimentally based calculations along with the introduction of a minimum of quantum mechanic theory brings to the student a new method of chemical analysis. The theoreticians and the organic synthesists are now able to relate their individual fields on a plane of computational and chemical understanding. Although the student may begin to understand what the computer can provide in terms of results, the actual decision upon which molecular modeling package to purchase should be made by the faculty and administration of the school.
There are numerous software packages for molecular modeling on the market. Among the numerous packages currently available are: Alchemy III(10), Nemisis(11), PC-Model(12), MacModel(13), Chem3D(14) and SPARTAN(15). Front end packages, like those listed above, provide an entry to the computational realm of chemistry. A comprehensive look at some competitive packages was published by Dr. Phil Bayes in 1989.(16) With the infusion of programs and packages available to schools and research groups, the number of publications supporting computational chemistry has multiplied as well.
The front end packages interface the three major components of molecular modeling. 1: Computer programs containing the parameters and equations designed by the computational chemists for molecular modeling generate the mathematical data and results. 2: A molecular builder/editor allows construction and editing of the molecule of choice. 3: Display of the numerical results as images is produced by a graphics processor.
Growing support from research increases the need for molecular modeling programs on college campuses. Middlebury College is in the midst of a growing trend in schools across the nation to have a molecular modeling laboratory. The two most common supportive arguments for introducing a molecular modeling lab are: research uses it, so our graduates must have exposure to it before reaching the work place(6) and the tool aids understanding too much to be ignored in a curriculum.(7) Middlebury has followed both of these issues: organic chemistry utilized portions of the Primer designed below, and a J-Term class will be offered specifically for molecular modeling and its application in advanced chemical processes.
SPARTAN provides a well rounded source for educational utility as well as top of the line research potential. The program is user friendly. It can be either be run by a three button mouse or through keyboard entry format. The user remains well informed of program functions. When calculations are submitted and completed the computer updates the user with text boxes. Warnings and notices also appear if a calculation is improperly setup or the calculation fails for some reason. SPARTAN also brings together a molecular builder/editor, a visual processor and calculational parameters all of which are able to inter-convert data and results. The builder has an extensive library of atoms and numerous possible valences for each atom, allowing any molecule to be built. The computational programs that are obtainable from SPARTAN's resources range from the most basic minimization calculation to higher ordered Ab Initio calculations. The visual display generates realistic rotations and translations of molecules, as well as a 3-dimensional viewing capacity.[*] Finally, the package is fast, powerful and capable of handling multiple tasks at one time. Other packages offer similar capabilities, but the SPARTAN software was most appealing to Middlebury because of cost and capabilities.
Many of the molecular modeling packages are based upon three basic premises: first, the ability to run iterative calculations, second, the use an empirical force field (Molecular Mechanics), a parameterized estimation of the Schrödinger Equation based upon valence electron theory(Semiempirical), or a much more focused view of the Schrödinger Equation which deals with all of the electrons (Ab Initio) to solve for chemical properties, and finally third, the ability to relate the generated mathematical output into a visual display of the data.
In iterative calculations, the molecular structure is estimated and an energy for that geometry is calculated. The structure is altered, or perturbed, a small amount and the energy of the new structure is calculated. This process is repeated until the calculations converge. One problem in several of the available programs is that local minimums are reached and the computer generates an inaccurate energy reading.(17) Once a minimized structure is calculated, the user has to determine what other chemical information is needed from the computer.
The process for submitting a calculation in SPARTAN is straight forward. First, the molecule is be built in the Builder. Before leaving the Builder, it is usually minimized. The user saves the built molecule, quits the Builder and enters the Workspace. In the workspace the calculations, properties, surfaces, volumes, searches, and drivers are setup. The desired calculations are submitted to the computational programs and the user waits for the results. Upon completion, the user informed of the completion via an alert text box. The molecule, and its resulting characteristics, can then be viewed from the Workspace.
At the end of the building phase, most molecules are minimized. For some special cases this would destroy a set configuration so it is not necessary, but in general, minimization provides a starting point for the proceeding calculations. This process is the foundation of molecular mechanics calculations.
Molecular Mechanics calculations are used to calculate bond lengths, angles and dihedral angles based upon the parameterized values in the calculation program's data bank. In most modeling programs molecular mechanics calculations use Equation 1 to determine the total energy of the molecule.[*] Note that other parameters can be input to influence or justify other chemical traits.
Etotal = Estretch + Ebend + Etorsional + Eout-of-plain + Evan der Waals Eq. 1
As a computational chemist needs grow, for example, they need to know more about a molecule than some distances and angles, the necessary level of theory and computation increases. Semiempirical calculations, as the name implies, is based upon some empirical force field calculations based upon the valence electrons and molecular orbitals and some parameterized data sets.
The Semiempirical molecular orbital methods are innovative mixtures of experimental data and theoretical expectations. These sets of parameters are generally based upon different families of molecules. One theory may work well with one type of molecule, but very poorly with others. Dependent upon what type of molecule is under investigation, the correct Semiempirical method can be determined from available tables.(18) The elements H, C, N, O were parameterized by 1985.(18) This broadened the scope of calculations to several organic molecules.
Austin Method 1 (AM1) is widely used in the primer because of its ability to generate reliable geometries, charge distributions, spin distributions, heat of formations, dipole moments and account for hydrogen bonding. SPARTAN version 4.0, AM1 is parameterized for 17 elements: H, B-F, Si-Cl, Zn, Ge, Br, Sn, I. Because of this void in complete coverage of all elements further theory may be need by the user. Parametric Method Number 3 (transition metal) [PM3(tm)] is used in the Primer to complete some of the calculations on organometallic complexes which have Ti, Zr, Hf, Ta, Cr, Mo, W, Co, Rh, Ni, or Pd in them.
The primer presented below is done so because the were no computational molecular modeling aspects in the organic chemistry curriculum at Middlebury College. By utilizing the capabilities of the molecular modeling package SPARTAN, as well as other computer programs, the goal is to introduce computational chemistry and more specifically molecular modeling for the organic chemistry class on molecular modeling and computational chemistry.
The primer consists of 11 chapters. Each chapter focuses upon a different topic. There is one introductory chapter which provides an overview of molecular modeling and shows the appropriate setup for sample calculations. Nine chapters dealing with computational means of chemical investigations, one as introduction and one showing pictures of the movies produced for a closer investigation of the images. The primer is designed in a modular fashion so that each chapter need not be used sequentially. Topics range from examining bond lengths of simple hydrocarbons though the HOMO and LUMO overlap of a Diels-Alder pericyclic reaction.
Three educational platforms are used throughout the primer: visual demonstrations, such as movies and presentations, homework assignments and laboratory experiments. The primer was designed to coordinate with many of the texts available to the students.[*](19) The movies display a variety of molecular characteristics. The homework assignments are provided for reinforcement of organic chemistry fundamental patterns and specific examples. The computational laboratory experiments integrate the synthetic chemistry that takes place on the laboratory benches with computational analysis.
Computer generated movies allow a wide variety of molecular and chemical properties of molecules to be viewed during a lecture or on Macintosh computers on campus. A compilation of the images and movies are available to the students through the campus server.
The homework problems utilize SPARTAN to investigate the current topics of study for the class. The introductory assignments acclimate the students to the program. The results from molecular modeling calculations provide information upon which the students can draw hypotheses and conclusions.
The integration of computer laboratory time with "wet laboratory" time brings students into the molecular level of chemistry. No longer are the students adding milliliters of solution to milligrams of substance, but dealing with the visual aspects, kinetics and thermodynamics of chemical reactions. The goal is, as T. W. Hanks of Furman University writes, "helping students develop their own models of the microscopic universe."(3)
The visualization of molecular structures is an easy task for most professors and students who have taken a full year of organic chemistry. Understanding several chemical properties comes from recognizing molecular symmetry and conformation. The second year students who have a basic background of chemical properties should be provided with all the academic resources that are currently available in the chemical domain. Molecular modeling is a current educational technique that allows the students to visualize molecules, contemplate the computational results and determine the significance of the results with experimental data. The resulting product is a student with a clearer image of chemistry, both as a field of study and a potential field of research.
Materials
The hardware used in the Middlebury College molecular modeling laboratory, and therefore in the production of the Primer, is: five Silicon Graphics Indy Workstations with MIPS R4600, 64 MB memory, 24-bit screen, 3-button mouse, IRIX Version 5.3 along with 13 Power Macintosh 7100/80 AV with 33,016K memory, System Software 7.5.1, MacX Version 1.5 and a 1-button mouse.
The software used for the molecular modeling laboratory at Middlebury College was SPARTAN, Version 4.0.(15) Production of the primer's movies utilized both IRIX Movie Maker Version 2.1 and IRIX Movie Player Version 2.1.(20) A mixed media presentation system IRIX Showcase 3.3(20) aided in many of the visual demonstrations. The Power Macintosh computers are connected through the Ethernet and MacX(21) makes the Macintoshes compatible with unix workstations. Fetch(22) was used to convert movies from the unix stations to the Macintosh format.
Methods
The following paragraphs are presented in an effort to describe the origin of each chapter and the questions and discussions within them.
Chapter one is provided as an introduction to both molecular modeling and SPARTAN. The students are shown example calculations as well as resources available to them. The SPARTAN Tutorial was used in reference, for not only content, but layout as well. Similarities between the two are purely based upon the usefulness of the characteristics utilized. (Imitation is the highest form of compliment.)
Chapter two involves an investigation of simple hydrocarbons. Bond lengths, angles, as well as molecular energies are calculated and reported by the student.
One of the primary aims of molecular modeling programs is to provide conformational analysis of molecules. The bond lengths of the calculated molecules must be close to the actual value to generate realistic data. Using the fact that many molecular mechanics programs, including MM2 and MM3, use data from hydrocarbons for their parameterizations, the resulting calculated bond length is often accurate to hundredths of angstroms on simple hydrocarbons.
The second chapter is designed not only to reinforce molecular geometry, bonding strength, distance and hybridization, but to also orient the student with some of the basic skills needed to use SPARTAN effectively.
It is important to note that the first calculational chapter in other texts[*] resembles the primer offered here.(23) This is not a form of plagiarism, but simply educators thinking along common lines. Basic calculations are easily arranged because of their simplicity.
Assignment I: Bond Lengths of Differently Hybridized Hydrocarbons
Ege's text offered the information necessary to complete this chapter.(19) As will be the case in the other four assignments in this chapter the text Experiments in Computational Chemistry has a similar assignment. The process is one which offers such educational aid, and it is a simple enough discussion, that repetition among research groups is common.
Assignment II: Conformational Analysis: n-Butane and a Substituted Ethane
This laboratory experiment has been performed by numerous facilities across the nation.(23) Dr. Bayes of Saint Mary's College and Professor Nelson of Middlebury College offered the foundation.
Assignment III: Cyclic Hydrocarbons
This homework assignment is a less involved form of one presented in the Hehre text. By providing the students with an opportunity to calculate the effective ring strain, SPARTAN allows manipulation and calculation of a visible model of the ring.
Cyclic compounds contain units of saturation. Utilizing SPARTAN to investigate both sources of saturation and unsaturation, this assignment relates the similarities of a chained alkane with its cyclic counterpart.(23)
Assignment IV: Relative Stabilities of Chair and Boat Cyclohexane
This assignment was a branch of the movie outlined below. The Hehre text (23) includes a similar assignment, but the accompanying movie and vibrational analysis is not included. A discussion on the SPARTAN e-mail account Spartanlist aided the clarity of the directions.(24)
Chapter three uses SPARTAN's ability to calculate dipole moments and the discussion in Ege(19) to generate the activities for the student.
Assignment I: Intermolecular Forces
The SPARTAN Tutorial predicated this assignment. By introducing the solvent effect calculations, SPARTAN allows hydrogen bonding to be investigated.
Assignment II: Intramolecular Forces
The investigation of a 1-2 diol originated from discussion with Dr. Professor Nelson and an assignment that she produced for the class.
Chapter four stemmed from the SPARTAN Tutorial(15) and the Ege text.(19) The tutorial provided insight to the possible investigations of dipole moments.
Assignment I: Dipole Moments of Solvents
The polarities of solvents were investigated through the Property option of SPARTAN. This assignment was generated by me.
Assignment II: Carbon Monoxide
Ege's text asked a homework question about the resonance structures of carbon monoxide and the relative dipole character or each structure. This assignment is a logical extension of the question posed.(19)
Chapter five, "Nomenclature", deals with stereocenters. These specific molecules were chosen due to SPARTAN's ability to manipulate the molecules on the screen.
An article from the New York Times was given to me by Professor Nelson. A brief homework was generated by having the students draw and name the two diastereomers of Albuterol. Other stereocenters were added to lengthen the assignment. Molecules of interest were taken from Ege.(19)
Chapter six was the extension of chapter 20 in the Williamson's text. "1-Butene, cis- and trans- 2-Butene from 2-Butanol."(17)
The Williamson text offers a chapter entitled Alkenes from Alcohols. Thermodynamics provides that the percent yield of products should be directly related to the stability of each possible alkene. Therefore, a study on the heat of formation is done to show students the relationship between Gas Chromatographic spectra and the stability of the products.
Chapter seven holds the widest array of possible portions; one movie, one lab and one homework assignment.
Assignment I: The Production of Adamantane
This movie is an animation that was suggested by Jeffrey Byers with the chemistry worked out in the Fleming text.(25)
Assignment II: Ferrocene Acylation
The Williamson text shows the synthetic technique necessary to acylate the ferrocene complex.(17) Computational analysis was also suggested by Kenneth Williamson, and were altered by Stewart Williamson.
Chapter eight is a one assignment investigation of tautomers.
Assignment I: Keto-Enol Equilibrium - Favoring the Enol
Professor Nelson suggested an equilibrium system that favored the enol form over the ketone form. The Lowery and Richardson text(27) provided a table of [enol]/[ketone] proportions in compounds. The phenol system was calculated from [Delta]Gdeg. estimated by J. B. Conant and G. B. Kistiakowsky.(28)
Chapter nine explores the vibrational calculations of SPARTAN through the Frequency Property.
Assignment I: Normal Modes of Vibration
The carbonyl stretching frequency is well founded experimentally in the literature. Ege's text offered sample questions for differing molecules.(19)
Chapter ten deals with Diels-Alder reactions. Electronic mail from Dr. Bayes and discussion with Professor Nelson lead to the two movies and the one in class demonstration.
The movies made were all created by the same process. Matthew Sontum and Dr. Bayes offered the technical support to save the pictures, generate the movies, and then transfer them to Macintosh computers through the Ethernet network. A technical appendix to this paper (Appendix A:Making Movies) and to the Molecular Modeling Primer (Primer Appendix B: Making Movies) is included with both movie making and movie displaying instructions.
The following paragraphs offer brief synapses of each movie and what went into their creation. They are offers here in order of appearance of the Primer.
Conformational Rotations of Butane along the C2-C3 bond
Several calculations have been done to show the conformational strain of the 1,4 carbon interaction.(23) Dr. Bayes provided some input in generating the rotation of n-butane.
Adamantane from Norbornane: Multi-step Reaction-Animated Chalk Board
Jeff Byers first suggested the possibility of showing a rearrangement of carbocation and hydrides. More specifically, he suggested the rearrangement of norbornane to adamantane from Fleming's Selected Organic Syntheses.(25) Three structures are shown simultaneously to clearly show the transformation of norbornane to adamantane: the molecular structure, the carbon structure and the electrostatic potential plot. By showing these three structures the audience is allowed more views of what is happening during the transformation.
SN2 reactions
These two movies show displacement of the leaving group and a five coordinated transition structure. Energy plots can be placed next to the reaction pathway to show the thermodynamic drive of the reaction. Showing transition structures is drawn in several text books, animating this reaction the logical continuation of these drawings. Both were created due a discussion with Professor Nelson.
Water Vibrations
SPARTAN's ability to calculate vibrational frequency's provided several frames of the three molecular vibrations. These three movies show the three normal modes of vibration of water. The decision to animate this vibration for CH241 was made by Professor Nelson.
Diels Alder Reaction: "Cracking and Dimerization of Cyclopentadiene"
Cycloaddition reactions, like the dimerization of cyclopentadiene, may be misinterpreted by students. By showing a well established concerted reaction pathway,(19) the movie provides a view of the production of the six membered ring. The cracking of the dimer was also made into a movie. The cracking movie was produced to enforce the reaction that the students perform in the lab. Even though the dimerization is the chemically favored product, the cracking reaction is one which provides the reactive species of cyclopentadiene.
Motion of Cyclohexane from Boat to Chair w/ Energy Plot
The Spartanlist e-mail group discussed such a movie. The energy plot shows the thermodynamic drive of chair favored conformation.
Computational analysis can be run along side several chemical reactions done at a laboratory bench. Two procedures were generated and one was tested by the organic chemistry class this fall. Williamson's text (17) offers several opportunities for computational exploration. The students are asked to write a report on the comparative values, structural difference and draw conclusions about the computational chemistry involved in the laboratory.
The alkene from alcohol lab was a direct extension of the suggestion of Williamson. The IR investigation is meant to coincide with the use of an IR spectrometer and was designed with Ege(19) as a reference.
Results
The molecular modeling primer is a tested collection of homework assignments, laboratory experiments and visual demonstrations. Many other applications of SPARTAN for organic chemistry were completed. The SPARTAN Tutorial (15) was completed in full to get a general idea of the limitations and capabilities of the program. Assignments given to Janet Nelson at molecular modeling conferences were also completed as introductory processes.
In terms of volume produced, fourteen homework assignments, six movies, one in class demonstration, and three laboratory experiments were generated. All of the homework assignments and laboratory experiments were checked for computational feasibility and application. The results have been saved in the unix workstation file under the file people/stewart/Primer/Answers. Numerical answers are provided in the teacher's edition of the primer, as well. Two of the homework assignments in the primer were completed in class. Real assignments completed by CH241/242 students are also included for both of them as reference. Five of the movies were shown to the Organic Chemistry class during lecture or pre-lab. One of the laboratory experiments was completed by the students in the academic year. A copy of a typical laboratory report and laboratory notebook is included as reference. Appendices that cover both the general overview and more specific interests is included as well.
Discussion and Conclusion
The molecular modeling primer is intended to introduce students to the computational capabilities of a molecular modeling package, like SPARTAN. Three educational tools are used in the primer: homework assignments, laboratory experiments and lecture demonstrations. The visual demonstrations were incorporated to introduce either motions of molecules in space and teach the students about chemical properties. The laboratory experiments were designed to allow hands on application of molecular modeling. The assignments were written as take home lessons on specific examples.
The numerous homework assignments cover a large range of molecular modeling topics. Strain energies, heats of formation, molecular orbitals and charge distributions are all utilized for investigation. Not only are the calculations run, but the students must understand at least some of the theory behind the calculation. Unfortunately, the computers may be used as a black box of calculations. However, the results of many calculations can be understood with some inspection.
The two laboratory experiments explore a process that the students can carry out at the bench top. The calculations involve basic principles, but a much broader scope of relationships are investigated.
Movies are available on the server for lecture demonstrations and the classroom presentations. They relay chemical information, reaction pathways, and internal motions of the molecules. They are available to the student and the teacher over the server.
SPARTAN has some limitations, there are computational links that cannot be ignored. Limits on the parameterized groups within the computational method used and the computer time needed to calculate all of the classes computations. Transition metals are now becoming more accurately parameterized,(18) but links are still missing. This is not meant to belittle SPARTAN, simply point out some of the subtle problems that limit its use.
Occasional specific examples of inconsistent relationships between the literature and calculations show how the mathematical nature of iterative processes. By displaying the local minimum, rather than the natural, or lowest, minimum, the students are forced to understand how the computer estimates, perturbs and generates the molecular results.
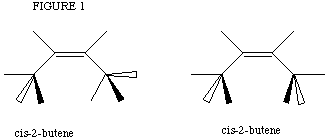
The lab "Alkenes from Alcohols: Dehydration of 2-Butanol" provides a perfect
example of such problems. The pi system in the butene products were drawn
numerous ways, until the lowest energy conformation was determined. Even
though Geometry Optimization calculations were run, the computer did not always
converge on the lowest energy. In Figure 1a cis-2-butene is shown with
its terminal hydrogen atoms at a calculated lowest minimum. If their symmetry
is disturbed before the calculation is submitted, the resulting conformation
may be Figure 1b. A Geometry Optimization calculation on this structure
results in a local minimum being found and higher conformational energy
presented as a result.
(a) (b)
There were other such cases of local minimum energies found by the iterative process. Other molecules include: compound X,[*] acylated ferrocene, numerous diketones and carbon dioxide. The simplest means of obtaining the lowest minimum is to re-submit the same molecule from different starting geometries. The lowest energy determined has a higher likelihood of actually being the lowest energy conformation. This is particularly the case when different starting materials converge to the same final conformation.
The overall performance of the computer system is worthy of praise. The networking throughout five Silicon Graphics allows five groups of students to operate at the same time. If more students need to access SPARTAN, the Ethernet connection to the Power Macintosh computers thirteen more computers are on-line with the one SPARTAN copy. Although there is a slight decrease in resolution, and the speed of the Macintoshes are much less than the Silicon Graphics, they perform the necessary tasks with time.
The primer is the one of the many preliminary steps in producing a functional molecular modeling lab. Students can either receive the whole primer, or individual portions can be copied and handed out. The applications are in their beginning stages, added assignments and modifications are welcomed at any stage. Teaching tools that do not function well provide minimal use to students. Therefore, alterations are welcomed an maybe even necessary. Because the workload associated with organic chemistry classes is often high, the introduction of new material is often difficult to fit in. The primer offers both a tutorial on molecular modeling and references toward texts which may provide insight for the students.
Appendices: A- Making Movies
The movies were made by a six step process which was first designed by Matthew Sontum. The process was as follows: make the molecule and all the necessary configurations of the molecule for a movie with SPARTAN, use the Snapshot desktop accessory to save the positions of the molecules as .rgb files, make the movie by placing successive picture after each other with the program MovieMaker, "Fetching" the movie onto the a Power Macintosh 7600 through the Ethernet, and finally viewing the assembled movie on the Macintosh computers.
To transfer all of the movies from the Silicon Graphics Workstations to the Macintosh computers and the Ethernet, the following procedure was followed:
1: Select "Movie Export Options" under the File menu sub-menu "Export".
2: Choose Apple Quick Time and Apple Animation [RLE] as Compression
3: Select "Export > Movie File..." The movie was then taken from the Silicon Graphics to the Macintosh computers by the Macintosh program Fetch. Once Fetch was opened the following directions were followed:
4: Enter into the text field that first appears in Fetch:
Host: Alta1
User ID: (e.g. stewart)
Password: my password
Directory: left blank
File Type : Binary
5: Find the desired movie file from the unix directory.
6: Click the Get button. This will bring up a text field where specific Type and Creator can be established. For all the movies transferred the Type was "MooV" and Creator was "TVOD". The movie was then saved into the Ethernet by placing it into the file Teaching Resources/CollaborativeProjects/Chemistry/ Faculty/ CH241/ CH241-Movies.
B - Methods of Calculations
i. Molecular Mechanics
Molecular Mechanics calculations use Equation 1 to determine the total energy of the molecule. The parameters for each of the energy terms in a molecular mechanics calculation may vary between individual front end packages, but Equation. 1 is the basic form for minimization and molecular mechanics calculations.
Etotal = Estretch + Ebend + Etorsional + Eout-of-plain + Evan der Waals Eq.1 Other terms are used to enhance the coverage of the Molecular Mechanics calculation. As an example, Eelectrostatic is occasionally used to include the electrostatic potential of each atom within the molecule.
The equations for determining the individual energy values are also in
standard form throughout several programs. Estretch is the energy
needed to stretch or compress a bond. Chemically, this parameter is very
high. The force constant,
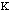 ,
in Equation 2,
,
in Equation 2,
Estretch =
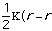 o)2
Eq. 2
o)2
Eq. 2
is very high. The variable
 is the bond length and
is the bond length and
 o
is the equilibrium value for the molecule.
o
is the equilibrium value for the molecule.
The energy to bend a bond, Ebend , is not as stringent as the
stretching constraints, but allows for minor movement in the molecule. The
force constant,
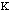 ,
in the Equation 3:
,
in the Equation 3:
Ebond =
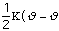 o)2
Eq. 3
o)2
Eq. 3
is moderately ranged. The angle between the atoms in a molecule is represented
as
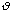 .
The equilibrium angle is represented by
.
The equilibrium angle is represented by
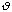 o.
o.
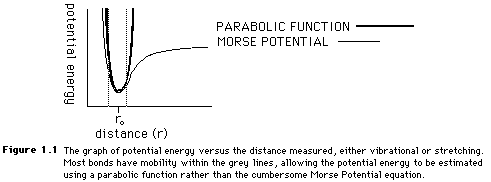
Both the stretching and bending values can be estimated with a parabolic function because of the similarities between the Morse Potential plot of potential energy and the parabolic functions. Figure 1.1 shows the overlap of the two curves. In Figure 1.1, the dotted lines show the average range of bonding stretches. Because the mobility of many molecules is limited, the parabola is a fairly accurate means of obtaining a Morse Potential energy value.
The energy for torsional angle adjustment is calculated from: Etorsional
=
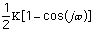 .
The force constant,
.
The force constant,
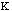 , is very low allowing for free rotation of bonds. This is due to the freedom
that compounds experience in rotations. The variable
, is very low allowing for free rotation of bonds. This is due to the freedom
that compounds experience in rotations. The variable
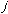 has the value of 2 or 3 depending upon the molecules symmetry. The torsional
angle,
has the value of 2 or 3 depending upon the molecules symmetry. The torsional
angle,
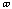 ,
is allowed to change for the calculations.
,
is allowed to change for the calculations.
Out-of-plane vibrational energy, Eout-of-plain, is determined by
the equation: Eout-of-plane =
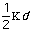 2.
The constant
2.
The constant
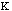 varies from system to system. In molecules with pi bond the constant is high.
The distance from the plane to the molecule of interest is represented by the
variable d.
varies from system to system. In molecules with pi bond the constant is high.
The distance from the plane to the molecule of interest is represented by the
variable d.
The final parameter to be discussed here for molecular mechanics approach to
molecular energy is the van der Waals energy term, Evan der Waals.
This term evaluates the attractive or repulsive forces between atoms. The
equation Evan der Waals =
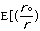 12
12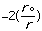 6]
uses the equilibrium distance, ro, the calculated
distance between atoms, r, along with the van der Waals potential of the
interacting atoms,
6]
uses the equilibrium distance, ro, the calculated
distance between atoms, r, along with the van der Waals potential of the
interacting atoms,
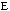 ,
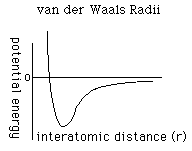
to determine the repulsion or attraction between atoms. Displayed in Figure
1.2 is the relationship described here, a graph of potential energy versus
inter-atomic distances, more commonly known as the Morse potential. The
equilibrium distance occurs at the minimum of the curve.
,
to determine the repulsion or attraction between atoms. Displayed in Figure
1.2 is the relationship described here, a graph of potential energy versus
inter-atomic distances, more commonly known as the Morse potential. The
equilibrium distance occurs at the minimum of the curve.
FIGURE 1.2
The above description of a calculation was for molecular mechanics. The force constants, bond lengths, angles and dihedrals are all based upon experimental data. X-ray crystallography provides much of the information for these parameters. As a computational chemist needs grow, for example, they need to know more about a molecule than some distances and angles, the necessary level of theory and computation increases. Semiempirical calculations, as the name implies, is based upon some empirical force field calculations based upon the electrons and molecular orbitals and some parameterized data sets.
ii. Semiempirical
The Semiempirical molecular orbital methods are innovative mixtures of experimental data and theoretical expectations. These sets of parameters are generally based upon different families of molecules. One theory may work well with one type of molecule, but very poorly with others. Dependent upon what type of molecule is under investigation, the correct Semiempirical method can be determined from available tables.(18)
The parameters for each method are determined from experimental data. This data allows the program to estimate certain difficult integrals from the Schrödinger time-independent equation. These estimations give results that follow the experimental rather than the theory. They also greatly decrease the computer time.
The introduction of the Neglect of Differential Overlap (NDO) Theory of the 1960's brought Semiempirical theory to molecular modeling programs. The Austin Method 1 (AM1) is used for calculations throughout the tutorial.
A discussion of a preliminary Semiempirical method, Modified Neglect of Diatomic Overlap (MNDO), is necessary to comprehend the expansion that the AM1 and PM3 methods offer. The paper published by M. J. Dewar and W. Theil in 1977 offer this objective: "The primary objective of the work reported in this series of papers has been the development of a quantitative treatment of molecular properties accurate enough, reliable enough, and sharp enough to be of practical value in chemistry, in particular in areas where experimental data are lacking or where current experimental procedures fail."(30) The goal was to provide more complete range for calculations and to improve what was currently available.
As calculational methods and models grew in complexity, some quantum mechanical problems arose. Approximations neglected electron repulsion integrals involving a one-center overlap, thus providing occasionally erratic results. Earlier methods combined 22 bicentric integrals into one estimated integral. Still other methods assumed that relationships between one centered integrals hold throughout the molecule.
The MNDO Theory, therefore, included key improvements in the aspects of molecular orbital discussion and calculations. The most important change being that the total energy of the molecule, Etotmol, became the sum of the electronic energy, Eel, and the repulsion, EABcore, between the atoms A and B.(33)
The mathematics involved in calculating the EABcore value are estimated from experimental results. This fact allow researchers the freedom to manipulate the parameter set to fit their experimental designs. If a trend is suspected in a family of molecules, experimental data can be input for the calculations and the math, being based upon experimental results, will show conclusive results. Those molecules that fit the proposed pattern give reasonable results, while those that might appear in the same category, but are actually in a different class of molecules, show dramatically different results. Certain small deviations in bonding energies, bond lengths or heats of formation occur because of the experimental results fusing with theoretical expectations.
The elements H, C, N, O were parameterized by 1985.(18) This broadened the scope of calculations to several organic molecules. The parameters were determined by a nonlinear least-squares optimization process based upon an algorithm devised by Bartels.(31) At this point the mathematics and computations become excessive and the mathematical texts in the references are offered for further reading. The methods were built upon the growing experimental data as well as the theoretical growth of the quantum mechanics. MNDO Theory rests as a primary set of parameters for molecular orbital methods of calculation.
With the understanding that calculations are based on experimental data and mathematical theory, AM1 and PM3 offer parameteric improvements. One of the larger problems of the MNDO theory was its neglect of hydrogen bonding. For better results, a new term had to be added. Simply changing the parameters would not suffice because the MNDO theory had not accounted for such interactions in any way. AM1 was created to be a stepping stone in producing the ideal method. It, however, provides consistent calculation of for many of the molecules.
AM1 is widely used in the primer because of its ability to generate reliable geometries, charge distributions, spin distributions, heat of formations, dipole moments and hydrogen bonding. As of 1990, AM1 is parameterized for 12 elements. Because of this void in complete coverage of all elements further theory may be need by the user. PM3(tm) is used to complete some of the calculations on organometallic complexes.
iii. Ab Initio
The third possible theory in calculation is the Ab Initio or first principles theory. The calculational programs now have the capacity to accurately estimate the integrals involved in the time-independent Schrödinger Equation:
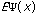 =(-h2
=(-h2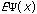 /
2m) (d2
/
2m) (d2
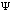 (x)/dx2)
+ U(x)
(x)/dx2)
+ U(x)
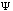 (x).
Eq.4
(x).
Eq.4
These calculations now deal with electrons and their position relative to their velocity. The basic premise behind Ab Initio calculations is this: it uniformly applies a continuous and unbiased field to molecular systems giving energies and kinetics of reactions.(35) Because these methods are not involved in any part of the primer, suggested readings for Ab Initio calculations are included as reference.[*]
Calculations can therefore be placed into three categories. Molecular Modeling, Semiempirical, or Ab Initio. It is imperative that the student understand the results that the computer generates. How the answers were generated is of importance as well, but the final answer must be understood to gain knowledge from the exercises.
References
(1) Krieger, J. H., Chemical & Engineering News, 1996, 74-17, 10.
(2) Pakarinen, J.; Smith, R.; Vainiotala, P; Pakkanen, T.; Kuttämaa, H. J. Am. Chem. Soc., 1996, 118, 3914.
(3) Hanks, T. W. J. Chem. Educ., 1994, 71, 62.
(4) Allinger, N. ;Geise, H. ; Pyckhout, W.; Paquette, L. ;Gallucci, J. J. Am. Chem. Soc. 1989, 111,1106.
(5) Engler, E. M.; Andose, J. D.; Schleyer, P. v. R. J. Am. Chem. Soc., 1974, 95, 8005.
(6) Ringan, N. S.; Grayson, L. J. Chem. Educ., 1994, 71, 856
(7) Casanova, J. J Chem. Educ. 1993, 70, 904
(8) Box, V.G.S. J Chem. Educ. 1991, 68, 662.
(9) Dr. Janet Nelson's CH453, Spring 1996, Middlebury College.
(10) Tripos Associates, Inc., 1699 South Hanley Road, Suite 303, Saint Louis, MO 63144.
(11) Oxford Molecular, Terrapin House, South Parks Road, Oxford, UK OX1 3UB.
(12) Serena Software, Serena Software, Box 3076, Bloomington, IN 47403-3076.
(13) Instar Software AB, Research Park IDEON, S-22370 Lund, Sweden.
(14) Cambridge Scientific Computing, Inc., 875 Massachusetts Ave., Suite 41, Cambridge, MA 02139.
(15) SPARTAN, SGI Version 4.0 GL, IRIX 5.2 Copyright 1991-1995 by Wavefunction, Inc., 18401 Von Karman Ave., Suite 370, Irvine, CA, 92715.
(16) Bayes, J. J. Chem. Educ. 1992, 69, 209.
(17) Williamson, K. Macroscale and Microscale Organic Experiments, (DC Heath, Massachusetts, 1994.
(18) Reviews in Computational Chemistry, Ed. K.B. Lipkowitz: D.B. Boyd, Vol. I, II, III , 1990, VCH, .
(19) Ege, S. Organic Chemistry, 3rd Ed., DC Health, Massachusetts, 1994.
(20) Silicon Graphics, Inc, 2011 N. Shoreline Blvd, Mountain View, CA 94039-7311.
(21) AGE Logic, Inc, 12651 High Bluff Dr., San Diego, CA 92130.
(22) Software Sales, Dartmouth College, 6028 Kiewit Computation Center, Hanover, NH 03755.
(23) Hehre, W.; Burke, L.; Shusterman, A.; Pietro, W. Experiments in Computational Chemistry, Wavefunction, Inc. 18401 Von Karman Ave., Suite 370, Irvine, CA 92715, 1993.
(24) Subj: SPARTAN: TS of Chair to Boat cyclohexane From: "Wayne Huang" <huang@mazda.wavefun.com> to: sparlist@mazda.wavefun.com
(25) Fleming, I.Selected Organic Syntheses, John Wiley & Sons, NY, 1977.
(26) Fox, M.; Whitesell, J. Organic Chemistry, Jones and Bartlett, Boston, 1994, p. 381.
(27) Lowery, T.; Richardson, K. Mechanism and Theory in Organic Chemistry, 2nd Ed.,Haper and Row, New York, 1981.
(28) Conant, J.; Kistiakowsky, G. Chem. Rev., 1937, 20, 181.
(29) Asperger, S.; Hegedic, D.; Pavlovic, D.; Borcic, S. J. Org. Chem., 1972, 37, 1745.
(30) Dewar, J.; Thiel, W. J.Am. Chem. Soc. 1977, 99, 4899.
(31) Bartels, R. University of Texas Center for Numerical Analysis, 1972, Report CNA-44, Austin,Texas.
(32) Stewart, J. J. Comput. Chem., 1989, 10, 209.
(33) Clark, T. A Handbook of Computational Chemistry; John Wiley and Sons, New York, 1985.
Suggested Reading - Reviews in Computational Chemistry, Ed. K.B. Lipkowitz: D.B. Boyd, Vols. I, II, III ,VCH, 1990. - A SPARTAN Tutorial, Wavefunction, Inc., Irvine, CA, 1993.
- User's Guide, SPARTAN Version 4.0, Wavefunction, Inc., Irvine, CA, 1995.
- Clark, T. A Handbook of Computational Chemistry; (John Wiley and Sons, New York, 1985).
- Hehre, W.J.; Radom, L.; Schleyer, P.v.R.; Pople, J. A. Ab Initio Molecular Orbital Theory John Wiley & Sons, New York, 1986. - Pople, J.; Beveridge, D. Approximate Molecular Orbital Theory, (McGraw-Hill, NY, 1970)
- Turner A. Methods in Molecular Orbital Theory, Prentice-Hall, NJ, 1974. - Williamson, K. L. Macroscale and Microscale Organic
Experiments, 2nd Ed.,(D. C. Heath & Co., MA), chapter 16.
[*] The 3-D viewing capacity is an overlap of a blue and red image viewed through red and blue lens glasses which come with SPARTAN version 4.0.
[*] See Appendix B: Calculations for a more complete description of all three calculational methods.
[*]The primary text in the 1995-6 academic year was the third edition of S. Ege's Organic Chemistry.
[*] most notably W. J. Hehre, L. D. Burke, A. J. Shusterman, W. J. Pietro Experiments in Computational Chemistry, Wavefunction, Inc. 1993.
[*] Williamson's text (17) offers thesynthesis of an unknown molecule entilted compound X.
[*] See References : Suggested Reading.