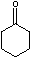 the equilibrium constant Keq is 4.1x 10-6 for [enol]/[ketone].
the equilibrium constant Keq is 4.1x 10-6 for [enol]/[ketone].
Many class texts available to the students coordinate with this primer. Each of the 11 chapters focuses upon a different topic. This, the first chapter provides an overview of molecular modeling, and will show you the appropriate setup for several example calculations. The primer is designed in a modular fashion so that each chapter need not be read or used sequentially. Topics range from examining bond lengths of hydrocarbons through the conformation of structure for a multi-step synthesis.
Three educational platforms are used throughout the primer: lecture demonstrations, homework assignments and laboratory experiments. The lecture demonstrations include both overheads and movies which display a variety of molecular characteristics. The homeworks are provided as expansions of other general organic chemistry material. The computational experiments brings the synthetic chemistry of a laboratory bench to computational analysis. A compilation of the images and movies are available to the students through the campus server.
Organic chemistry students with basic background to chemical properties should be provided with all the academic resources that are currently available in education. Molecular modeling is as up-and-coming academic technique that allows students to visualize molecules, interpret experimental results and determine the significance of the results when compared with experimental data. The resulting product is a student with a clearer image of chemistry, both as a tool for study and an instrument in research. To the Student
The goal of this primer is to introduce you to a new realm of chemistry that is quickly becoming a tool in education, research and industry: molecular modeling. As you use this book in your studies there will be times when the material covers only the surface of a problem. There will be times when the projects cover beyond the text used in class. Again, our aim is to teach both the simplicity and complexity of molecular modeling.
The opportunity to use the computer program SPARTAN 4.0 was introduced in the summer of 1995 and since then has become a focus of new studies in organic chemistry at Middlebury College. There are numerous articles available utilizing molecular mechanics in both the classroom and in research groups.[*] You should find the program very user friendly and its graphical displays easy to visualize.
A brief introduction to some of the vocabulary of molecular modeling is needed at this time. Molecular modeling is a theory of calculation for determining the geometric structures of molecules based on a Newtonian physics, empirical data, and mathematical theories.
The molecules of interest are drawn within a Builder. The molecules are then placed into a Workspace where the different calculations are set up and submitted. Most of the calculations run are done by a computational device through an iterative process. The molecular characteristics are perturbed slightly numerous times until the calculations converge to a minimum. The images and results are manipulated by a graphics driver that create the pictures.
There are three general types of molecular modeling calculations that can be done on a molecule: Molecular Mechanics, Semiempirical, and Ab Initio.*** All three have varying methods which correspond to different theory of calculations. The commercially available methods are numerous, each with their own strengths. Throughout this text four different task sets will be used: MM2, MM3, AM1 and PM3(tm). MM2 and MM3 are Molecular Mechanics task calculations, while AM1 and PM3(tm) are Semiempirical task calculations.
Molecular Mechanics calculations are based upon the Newtonian physics of molecules. Masses, bond strengths, inter-atomic distances, electrostatic potential, van der Waals forces, and other forces are all taken into account. These calculations are all based upon experimental. None of the Molecular Mechanics calculations take delocalized electronic effects into accounts.
The Semiempirical molecular orbital methods are innovative mixtures of experimental data and theoretical expectations. These sets of parameters are generally based upon different families of molecules. The parameters for each method are determined from experimental data. This data allows the program to estimate certain difficult integrals from the Schrödinger time-independent equation. These estimations should provide results that follow both the experimental values and theoretical expectations.
Ab Initio, or first principle, calculations are based solely on mathematical quantum mechanical theory. The estimation of integrals and energy terms is taken to a higher level of involved calculation in Ab Initio theory. For this, and other reasons, Ab Initio theory is not used in the primer.
Minimization, which takes place directly after most molecules are built, is a low level Molecular Mechanics calculation. It provides a starting geometry and strain energy for the molecule. The minimization calculation is an empirical molecular mechanics calculation based on the SYBYL force field.(1)
The Semiempirical method AM1 is the most commonly used molecular calculation in the primer. In SPARTAN version 4.0, AM1 is parameterized for 17 elements: H, B-F, Si-Cl, Zn, Ge, Br, Sn, and I. Because of this void in complete coverage of all elements further theory may be need by the user. PM3(tm) is used to complete the calculations of inorganic or organometallic complexes which have Ti, Zr, Hf, Ta, Cr, Mo, W, CO, Rh, Ni, or Pd in them.
A Brief User's Guide
Use of the Three Button Mouse and the One Button Mouse
If you are at a Silicon Graphics Indy Workstation there will be a three button mouse. If you are at a Macintosh and you are using MacX, you will have to use the arrow keys to convert the one button mouse to a three button one.
CONVERSION FOR 1-BUTTON to 3-BUTTON MOUSE
SGI MacX
Left button ------> mouse button
Middle button ---> only left arrow
Right button -----> only right arrow
The left mouse button operates the File Commands.
NOTE- When using this table, push the keyboard button then the mouse button. (Ex. Shift then right mouse (arrow) to zoom)
Keyboard
nothing
picking
x/y rotate
x/y translate
space bar
space & control
spc, cntrl, shift
---
---
---
rotate bond
global rotate
z rotate
---
global translate
z translate
Acclimate yourself to moving the molecule with the mouse. Refer to the chart
for a one button mouse and a three button system when necessary. View the
molecule down various bonds and through planes of symmetry.
Left
Button
Middle
Button
Right
Button
shift
---
z
rotate
zoom
all
How to build molecules:
Select New from the File menu with the left mouse button. This will bring you into the Builder. There will be two screens present. The one on the left is the work space and is blank. The smaller one, on the right, has some hybridized atoms, a text box and some commands.
To build any molecule you must select the atoms by clicking, with the left mouse button, first on the appropriate symbol, then in the work space. Each yellow line seen on the atom represents an open bonding valence. To make a bond click on the atom to add and then on any yellow line with the same hybridization. For example, an sp3 C with an sp3 O, or an sp2 N with an sp2 C Any unfilled yellow lines will be replaced with hydrogens when the molecule is saved, so there is no need to add hydrogens.
After building the molecule, click the Minimize button in the command box (lower left corner of right hand box.) Minimization is not always necessary, but it provides a starting point for the computer. The computer applies a simple force field to the built molecule and places atoms in appropriate molecular geometries.
To Save a built molecule, select Save As from the File menu. Save the molecule by clicking in the text box and type "name of the molecule.yourinitials" (be sure not to leave spaces) Click save.
To Quit the builder, select quit from the File menu. The molecule will appear on the original SPARTAN screen.
Visualizations of Molecules:
Once a molecule is on the main screen open the Model menu and look at the different styles of molecular depictions. Select the type of view by clicking it with the left mouse button. The space filling model is of particular interest because it closely resembles the electron density of the molecule. Using the space filling representation continuously, however, slows the computer down; it is easiest to manipulate the molecules in either the ball and stick or tube models. (Note that the graphics of the Macintosh computers have much lower resolution than the Silicon Graphics Indy Workstation)
Making Measurements
Once a calculated molecule is on the main screen open up the Geometry menu and select Distance or Angle or Dihedral. Measure the distances around the molecule by clicking on either the bond between two atoms or on two separate atoms. To measure an angle, select three adjacent atoms by clicking once with the left mouse button. To measure any dihedral angle (the angle of rotation along a bond), select four adjacent atoms by clicking once with the left mouse button. The selected atoms will become yellow and the measurement will appear in the upper left hand corner of the screen. You can measure the distance any two atoms, bonded or not. The same is true for angles and dihedral angles. A data box will appear giving you the measurement in either Angstroms (10-10 meters) or degrees. NOTE: When taking any measurement the order in which the atoms are selected is important.
Setting Up and Submitting a Calculation
Select the molecule to calculate by clicking with the left mouse button once it is on the main screen. The title of the molecule will appear at the top of the SPARTAN screen. Open the Setup menu and select calculational theory you wish to use. This is usually Semiempirical or Molecular Mechanics.
NEVER run Ab Initio calculations unless instructed to by the professor, as they take up useful computer time.
A new text box will appear and you must now specify the calculation you wish to perform. If you would like to title the calculation, click with the left mouse button, inside the box to the right of the Title: prompt and type your title. It is not necessary to title a calculation. At the Task prompt click on the box and select the appropriate task, quite often it is Geometry Optimization. At the Level prompt click on the box and select the appropriate level of calculation, quite often this is AM1. Enter any options or solvent effects as instructed. The Charge and Multiplicity should also be entered (usually 0 and 1 respectively). The defaults are usually correct. With the left mouse button click Save after the correct calculation perimeters have been set up. This will return you to the main SPARTAN screen. You can now setup any Properties, Surfaces, or Volumes by selecting the proper choice under the Setup menu.
To submit the calculation and conformation search open the Setup menu again and select Submit. This will tell the computer to start calculating. Click OK when the text box "Your job 'molecule X.' is running" In a moment the calculation will be done and you will be notified. Click OK when the text box "Your job 'molecule X.' has completed."
Closing SPARTAN:
Open the File menu and select Quit. A text box will appear asking to close all molecules, if any were open, click yes. Quit MacX, as well.
The first assignment is provided as a brief refresher of hybridization and molecular geometry. Hybridization of molecular orbitals and geometric shapes of molecules are investigated. The second assignment could be reported by the student as a molecular modeling laboratory experiment. Two conformational investigations are calculated leading a discussion based upon sigma bond rotations and conformational stability. The last two assignments are based upon cyclic molecules and their chain counterparts.
Chapter 2: Hydrocarbons (#1) Name: ______________
Date: / /
Organic Chemistry I
In this assignment is you should acclimate yourself with the SPARTAN Work Stations. By constructing, manipulating, and minimizing molecules you should gain a more visual understanding of simple organic molecules.
Lewis Dot Structures and Valence Electron Shell Repulsion Theory should both be taken into account while you complete this assignment. Hybrid orbitals allow for different molecular structures. Generating the correct structure will be important in this assignment.
1) In the SPARTAN builder construct and minimize ethane (C2H6).
2) Save the molecule and quit the builder.
3) Setup and Submit a Molecular Mechanics MM3, Geometry Optimization Calculation.
4) Once the calculation has completed open the Geometry menu and select Distance and record the distance between the two carbon atoms below.
5) Build and Geometry Optimize ethene (C2H4) and ethyne(C2H2) and determine their carbon carbon bond lengths by the same process. Record your results below.
5) Draw accurate three dimensional representations of the three molecules below.
Molecule
|
Carbon
Hybridization
|
C-C
Bond Length (Å)
|
| ethane
.............................
|
________________
|
______________
|
| ethene
.............................
|
________________
|
______________
|
| ethyne
.............................
|
________________
|
______________
|
Draw ethane here:
Draw ethene here:
Draw ethyne here:
Chapter 2: Hydrocarbons (#2) Name: ______________
Date: / / Organic Chemistry I
and a Substituted Ethane
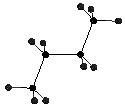
n-Butane has a sigma bond that rotates as one of the molecule's internal motions. The thermodynamic energy of the molecule varies at each position. With an non-substituted n-butane the position and energy levels are symmetric from 0deg. to 180deg. and 180deg. to 360deg.. Different dihedral angles give different heats of formation and therefore different internal energies.
In this assignment you are going to use a coordinate driving option on n-butane to calculate the heat of formation of 18 different conformations of n-butane. From the calculated energies a graph can be generated that relates dihedral angle to energy level.
In SPARTAN:
1) Build, Minimize and Save n-butane. Quit the builder.
2) To measure the dihedral angle open the Geometry menu and select Dihedral Angle. This will open a new window in which you should select the four carbon atoms in sequence (C1, C2, C3, C4).
3) A text screen "pops up" when you select four atoms. This screen tells you the current dihedral value. It should read very close to
+/- 180deg..
4) If the dihedral does not read within five degrees of +/- 180deg. click OK and select Edit Molecule under the Build menu. To twist any bond select it with the left mouse button so that it becomes a dashed line. Hold down the space bar and the middle mouse button at the same time and move the mouse up and down until the angle is close to 180 degrees. Save the changes and quit the builder.
5) A calculation must be done to find out the Heat of Formation of the molecule. Enter the Semiempirical setup screen by selecting it from the Setup menu with the left mouse button. Set up an AM1, Geometry Optimization calculation.
6) Open the Coordinate Driver option under the Build menu.
7) The molecule will appear in a new box with a text box on the right. In the text box select "Define Dihedral." Click on all four Carbon atoms in a row. Once four atoms are selected a new text will appear. Be sure you have chosen the four carbon atoms.
8) The new text box tells you the current dihedral angle and allows you to set up the appropriate rotations for the driver.
9) The text should be set to read the following...
From: +/- 180.00
To: 0.000 Steps: 18
Sigma: 100.00
10) Click save to quit the Coordinate Driving Option.
11) Submit the calculation and coordinate drive by clicking on Submit under the Setup menu.
12) Once the job is submitted confirm the action by clicking OK in the text box that appears.
13) After the job is completed, the computer will notify you when this happens, click OK in the text box. This will inform the computer to generate all 18 molecules into a file. Again, click OK in the new text box.
14) A new text box will appear with the n-butane molecule. To see the energies of each conformation select Show Energy under the View menu of the Coordinate Driver text box. The energy is in kcal/ mol. Select any one of the listed names to see the conformation on the screen. Feel free to animate the rotation as well.
15) Record the results and generate a graph of the energies versus the dihedral angle. Because the molecule's conformations are identical from 0deg.-180deg. and 180deg.-360deg., the graph can be duplicated for a full 360deg. rotation.
Part II... a substituted ethane
1) Build ethane in the Builder.
2) Choose a substituant to place onto one of the open valences on carbon. (for example: F, Br, OH, CH3)
3) Save your substituted ethane. Name it ethane.(your initials).0-180.
4) Quit the Builder.
5) Note which molecule is open. The title of the selected molecule appears at the top of the SPARTAN menu bar. Open the Coordinate Driver Option under the Build menu.
6) In the new text box select Define Dihedral and then select the substituted group, then the closest carbon, then the second carbon and finally the hydrogen that is in the plane of the substituent.
7) Make the necessary changes to the text box sop that it reads:
From: -180
To: 0 Steps: 18
Sigma: 100
8) Quit the Coordinate Driver (File menu) Save any changes made.
9) Choose Semiempirical under the Setup menu.
10) Setup an AM1, Geometry Optimization calculation.
11) Submit both the coordinate driving and the Semiempirical calculation by selecting Submit under the Setup menu. Click OK to confirm the calculation.
12) Click OK at prompts "calculation 'name' finished" and "Generating list of molecules"
13) Show the individual energy calculations by clicking, with the left mouse button, on Show Energy under the View menu of the Coordinate Driver text box.
14) Write down the energies and their corresponding dihedral angle. To measure the dihedral angle, select Measure Dihedral under the Geometry menu. It is important to follow the same pattern of selecting dihedral angle, substituent, carbon, carbon, hydrogen (be sure it is the same one)
15) Make a graph of the energies versus the dihedral angle in your note book. Discuss similarities and differences in the two graphs.
Chapter 2: Hydrocarbons (#3) Name: ______________
Date: / /
Organic Chemistry I
This assignment will determine the heat of formation and the bond angles of three cyclic hydrocarbons. The strain energy on the three rings can also be investigated by comparison to their linear counterparts.
1) Build cyclohexane (C6H12).
2) Save the molecule and quit the Builder.
3) Submit a Semiempirical AM1, Geometry Optimization to determine the heat of formation of the molecule. Confirm the submission by clicking OK in the text box that appears.
4) After the calculation has completed, open the Geometry menu and select Angle. Highlight three adjacent carbons on the six membered ring. Record the angle between the three carbon atoms below. Double check the five other bond angles.
5) Measure the hydrogen-carbon-hydrogen angle for each carbon.
6) Build cyclopentane (C5H10) and cyclobutane (C4H8) and determine their carbon-carbon-carbon and hydrogen-carbon-hydrogen angles by the same calculational process. Record your results below. Note that the C-C-C bond angles are different! Why?
7) Build a linear conformation of hexane (C6H14).
NOTE: if the builder minimizes the molecule into a carbon chain that is not in the same plane, redo the drawing.
8) Save the molecule and quit the builder.
9) Submit a Semiempirical AM1, Geometry Optimization to determine the Heat of Formation of the molecule.
10) Under the Geometry menu select Angle and select three adjacent carbons on the six membered ring. Record the angle between the three carbon atoms below. Check the five other bond angles, why are they different?
11) Measure the hydrogen-carbon-hydrogen angle for each carbon. How do these compare with the cyclohexane? Where are the differences the greatest? Why?
12) Build pentane(C5H12) and cyclobutane (C4H10) and determine their carbon-carbon-carbon and hydrogen-carbon-hydrogen angles
by the same process. Record your results below.
13) Submit a Semiempirical AM1, Geometry Optimization to determine the Heat of Formation of each molecule. Record the energy value in Table 1.
DRAW & REPORT BOND ANGLES BELOW:
Draw cyclohexane and label the bond angles here:
Draw hexane and label the bond angles here:
Draw cyclopentane and label the bond angles here:
Draw pentane and label the bond angles here:
Draw cyclobutane and label the bond angles here:
Draw butane and label the bond angles here:
TABLE 1
Molecule
|
Heat
of Formation (kcal/mol)
|
| cyclohexane
........................
hexane ...................................
|
______________
______________
|
cyclopentane ......................
pentane .................................
|
______________
______________
|
cyclobutane ........................
butane ...................................
|
______________
______________
|
Date / /
Organic Chemistry
Cylclohexane undergoes a flipping motion as a structural vibration. The ring strain induced by this flipping mode can be calculated by the molecular modeling program SPARTAN. The energy derived from Semiempirical calculations promotes this investigation.
Just as any transitional state motion has an energy of activation, the flipping mode goes through an energy maximum in order to complete the new conformation. Thermodynamics favors the conformation with the lowest energy value.
Building transition states on SPARTAN is a simple process of constructing the initial and the final conformations. The program then uses a linear synchronous to determine the best transition form. By saving this conformation and running a single point energy analysis you can see a filtered view of the flipping transition.
For this assignment you must draw an energy diagram for the conformational vibration, discuss what is happening to the molecular structure of cyclohexane and give a brief synapse of why one conformation is favored over another.
To Build Boat Cyclohexane:
1) Select the cyclohexane ring in the Builder. It is in the Chair conformation, save this as "chair(your.initials)"
2) Add a bridge methyl group
3) Connect bridge across the ring.
4) Minimize this strained complex.
5) Select Delete Atom from the right hand text box and delete the bridged methyl group.
6) Minimize again, save and quit.
----
To Build the transition form:
1) Open both chair and boat conformations.
2) Select Boat so that its title appears at the top of the SPARTAN Monitor.
3) Open Transition Search under the Geometry menu.
4) Click on the Chair conformation to indicate the Product. A new screen will open with both molecules on it.
5) Match each atom with its counterpart. Be careful with the hydrogens. You can rotate the molecule by moving the mouse in the screen with the middle button down.
6) Select Generate from the Edit file. This will generate a transition structure through a linear synchronous process. The product is the transition structure and a Semiempirical, AM1, single point energy of it can be submitted.
7) Compare the three molecules single point energies and draw a graph of the motions pathway.
Chapter 3: Hydrogen Bonding Name: _____________
Date: / /
Organic Chemistry I
The ability of hydrogen to stabilize a structure by absorbing electrons from electron rich areas in molecules is known as an intermolecular interaction. Structures gain stability through this dispersal of charge. Water has long been known to interact with some organic molecules. Here we will look at the stability added when a series of alcohols are calculated in solution and out of solution.
1) Build four of the following molecules: 1-propanol, 2-propanol, 1-butanol, 2-butanol, 1-pentanol, 2-pentanol, or 3-pentanol.
2) Save the four molecules twice. Once as "1-propanol.(your.initials) and once as "1-propanol.(your.initials).H2O
3) Submit a Semiempirical, AM1, Geometry Optimization calculation for all four molecules without the .H2O ending. (the solvent should be "none")
4) Submit a Semiempirical, AM1, Geometry Optimization, water solvent calculation, for all four molecules with the .H2O ending. (the solvent should be "water")
5) Compare and contrast the four molecules in terms of heats of formation.
Chapter 3: Hydrogen Bonding Name: _____________
Date: / /
Organic Chemistry I
1) Build 2,3 butanediol in at least three different conformations. Change the relative positions of the hydroxyl group, not the carbon carbon bond. To do this select the C-O bond so that it is a dashed line. Hold the space bar down and the left mouse button, while moving the mouse in a vertical motion. As you build various conformations, keep in mind the intramolecular forces active in this molecule.
2) Save each and quit the builder.
3) Submit a Semiempirical, AM1, Geometry Optimization calculation for the molecules.
4) Look at the heats of formation for each one, and determine the lowest conformation of 2,3 butanediol.
Certain molecules contain no dipole moment. Diatomic molecules, where the two atoms are the same, O2, N2, Br2, are a few examples, do not have a dipole moment. Symmetry about a center can also cancel out any charge imbalance. CO2, BF3, CCl4, are examples of these electronically balanced molecules.
The dipole moment is the sum of all the dipole vectors in the molecule. This is why CCl4, CO2, BF3, have no net dipole moment. However, once that symmetry is broken, such as in CH3Cl, the resulting dipole moment can be very strong.
The net dipole moment is shown by an arrow originating at the center of positive charge, and pointing towards the center of negative charge. Note that the center of charge does not need to be on an atom. It can, and quite often does, occur in between atoms of identical charge.
CHAPTER 4: Dipole Moments
Name: _____________
Date: / /
Organic Chemistry I
Solvents:
Possible molecules: dichloromethane, trichloromethane, tert-butyl chloride, dimethylsulfoxide (DMSO), acetone, or benzene.
1) Estimate the location of a dipole moment in the above molecules.
2) Use SPARTAN to confirm your predictions.
3) Build four of the listed molecules in the Builder.
4) Setup a Semiempirical, AM1, Geometry Optimization calculation. 5) Under the setup menu select properties. In the text box that appears click Dipole Moment and then click save.
6) Submit the calculation.
7) To view the position and value of the dipole moment select Properties under the Display menu. Then select the Dipole sub menu. The molecule selected will appear with a text box showing both the value and the coordinates of the dipole vector. The moment is the sum of all the electronic distributions calculated by SPARTAN.
8) Compare the calculated values with those in the literature. Note any differences between the calculated values and those cited in the literature.
9) Why are solvents like DMSO and acetone are used? What intermolecular factors must be taken into account when dealing with these solvents?
CHAPTER 4: Dipole Moments
Name: _____________
Date: / /
Organic Chemistry I
1) Build CO in the expert builder (enter the builder and select expert) and select the -X- bond hybrid. Build -C-O- and delete the open valences. Select the triple bond and double click the remaining C-O bond.
2) Save and quit the builder.
3) Setup a Semiempirical, AM1, Geometry Optimization calculation. 4) Under the setup menu select properties. In the text box that appears click Dipole Moment and then click save.
5) Compare this value to the value in the literature.
6) What provides this molecule with the decrease in dipole moment? What effects does this decrease have on carbon monoxide's stability and reactivity?
CHAPTER 5: Nomenclature Name: _____________
Date: / /
Organic Chemistry I
1) Build and minimize three of the following:
S-glyceraldehyde, R-glyceraldehyde, S-Albuterol, or R-Albuterol
2) Draw a representation of the stereocentered atom below. Label
the proper hierarchy of substituents.
By following the directions given by the Williamson text Macroscale and Microscale Organic Experiments(5) you will produce three alkenes from 2-butanol. SPARTAN will allow you to calculate the relative heats of formation of the products. Correlations, based upon some thermodynamic assumptions, can be made between these two experimental data sets.
CHAPTER 6: Elimination Reactions Name: _____________
Date: / /
Organic Chemistry I
Calculated heats of formation relate to the strength and stability of the molecule submitted. Final and initial energies can be compared. The dehydration reaction need acidic conditions and high temperatures to complete the reaction.
At the Lab Bench...
1) Follow the instructions in Williamson (3) Chapter 12.2 to make the three butene products.
At the Keyboard...
1) Enter the Builder by selecting New from the File menu.
2) Build ethene first. Select the sp2 hybridized carbon and click on the work space on the left. The carbon atom will be in a position which is difficult to see the open valences so, rotate the atom with the middle mouse button.
3) Ethene will be your starting point for all three products. First, build 1-butene. Place a methyl onto any valence of ethene. Now, add a second methyl onto the first methyl.
4) Minimize the structure
5) Save your molecule as (your initials).1-butene.
6) Quit the Builder.
7) Submit a Semiempirical, AM1, Geometry Optimization calculation.
8) Obtain the Heat of Formation of the molecule by selecting the Energy option under Properties option under the Display menu. Record this value in your Lab notebook.
9) Sketch 1-butene as a 3 dimensional image in your notebook. The location of hydrogens is important, so draw carefully.
10) Select New under the File menu and build cis-2-butene starting with ethene, the same as above. Add the two methyl groups in the appropriate positions with the double bond.
11) Minimize and save the molecule. Save this one as:
(your initials).cis-2-butene
12) Submit a Semiempirical, AM1, Geometry Optimization calculation.
13) Note the Heat of Formation of the molecule by selecting Energy under the Properties option under the Display menu.
14) Build trans-2-butene in a similar fashion as above. Build ethene first and add the two methyl groups in the appropriate positions with the double bond.
15) Minimize and save the molecule. Save this one as:
(your initials).trans-2-butene
16) Quit the builder and submit the same calculation as above. Note the Heat of Formation of the molecule.
17) Sketch each of the molecules in your lab book, noting the position of each atom.
18) Exit SPARTAN by selecting Quit under the File menu.
19) Find the heat of formation of each alkene in the literature. If the values are different than those you calculated, which are more accurate? How do the relative differences compare? If placed in order of highest to lowest energy, are the results the same from SPARTAN as from the literature?
The Synthesis of Adamantane
This movie provides a visual look at one of the possible pathways for the alterations that occur when norbornane looses a hydride. Adamantane is produced through this suggested sixteen step reaction. 1,2-Hydride shifts, 1,2-methyl shifts, carbonium ion rearrangement and hydride replacements are all processes involved in this reaction.
The proposed steps for carbonium ion rearrangement to form adamantane are(4):
00 - rotation of the hydrocarbon so that the initial conformation can be seen
01 - initiation by the s-butyl cation removing a hydride from one of the many possible positions of removal. Note it is an open hydrogen without too much steric hindrance.
02 - a 1,2 methyl bond shift relocates the carbocation to a secondary position. Note the strain on the quaternary carbon.
03 - a 1,2 hydride shift
04 - a 1,2 methyl shift relocates the carbocation to a secondary position.
05 - a multiple bond, or through space, hydride shift.
06 - a 1,2 hydride shift
07 - strained rearrangement with a 1,2 methyl shift
08 - rearrangement to ease strain no bonds are broken, bond rotations form two less strained six membered rings, one with a bridging methyl group
09 - a 1,2 hydride shift
10 - a 1,2 methyl shift, the molecule is becoming more global and symmetric
11 - a hydrogen transfer - can also be protonation and deprotonation from an s-butyl cation source and n- butane
12 - a 1,2 methyl shift
13 - final hydride replacement to form adamantane.
CHAPTER 7: Substitution Reaction Name: ______________
Date: / /
Organic Chemistry II
For this lab you are to first either obtain or synthesize ferrocene. Then, through the directions of Williamson Chapter 44 acylate the organometallic complex.
With SPARTAN investigate the geometry of the complex and its acylated product.
Enter the Expert builder and construct ferrocene by following these directions:
1) Select the -X- central hybridization along with Fe from the list of elements and click in the work space.
2) Click on the Ligand box in right hand text field and choose the cyclopentadienyl ligand.
3) Place at cp ring at the end of both Fe valences.
4) Constrain the cp-Fe-cp bond angle to 180deg.. To do this open the Constrain Angle sub-menu of the Geometry menu in the Builder. Select the point of attachment for one cp ring, the Fe atom and the second point of attachment on the second cp ring. The text box should have an angle reading of 180deg. and a sigma value of 100.00. If that is not the case, place those values into the appropriate positions. Click OK.
5) Minimize the structure. The complex should obtain D5d symmetry. If it does not, you must rebuild it.
6) Save the complex as "ferrocene.(your initials)" and quit the builder.
7) Setup and submit a Semiempirical, PM3(tm), Geometry Optimization calculation. The PM3(tm) model is used because it has been parameterized for transition metals (tm).
8) Once the calculation has completed save a copy of the complex as ferrocene.acylated(your initials) by choosing Save As under the File menu.
9) With the ferrocene.acylated(your initials) complex selected, enter the Edit molecule in the Build menu. This will place you in the Builder.
10) Acylate the complex on one of the cp ring's open valence.
11) Do not minimize this structure. Save this structure and quit the Builder.
12) Submit the same Semiempirical calculations as above. (It takes about ten minutes to compute, be patient)
13) Note the heats of formation of the two complexes. These values are not very helpful due to their great difference, however, note them in your lab book.
14) Observe the location of the carbonyl group, is it in or out of the plane of the cyclopentadiene ring? What does this fact imply about resonance stability?
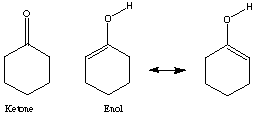
CHAPTER 8: Keto/Enol Tautomerism Name: ______________
Date: / /
Organic Chemistry II
The equilibrium of keto-enol tautomers usually greatly favors the ketone.
For example, in
the equilibrium constant Keq is 4.1x 10-6 for [enol]/[ketone].
Yet, the equilibrium constant, Keq, for
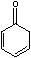 is 4x1013 for [enol]/[ketone].
is 4x1013 for [enol]/[ketone].
1) Why is the enol form favored so dramatically?
2) Use SPARTAN to determine the Heats of Formation of all four structures.
3) Build all four molecules and submit a Semiempirical, AM1, Geometry Optimization calculation.
4) From the calculated values of heat of formation for the molecules and the equations below determine the equilibrium constant Keq. (assume room temperature of 25deg.C)
[Delta]G = -RT lnK
[Delta]H= [Delta]G-T[Delta]S
5) Can we assume [Delta]S << 1?
Movies of the normal modes of vibration for water accompany this chapter as well.
CHAPTER 9
Infrared Assignment Name: _____________
CH 241
In SPARTAN:
1) Build all six molecules listed above.
2) Setup a Semiempirical AM1, Geometry Optimization calculation for each of the molecules listed above.
3) Select the Property (a sub menu of the Setup menu) of Frequency for each calculation.
4) Once the calculation is complete, you may observe the vibrations by selecting Frequency under the Display menu.
5) Determine which frequency is the C=O vibration in each molecule. In the case of 2,4-hexanedione be sure to predict which peak refers to which C=O vibration.
6) Compare and contrast:
A: acetone and formaldehyde,
B: cyclohexanone with cyclobutanone,
C: the two frequencies of 2,4-hexanedione.
Butadiene and Acrolein Discussion
Butadiene and Acrolein Discussion-
Which product will form? The endo or the exo?
1) Observe the overhead of HOMO and LUMO overlap of acrolein and butadiene.
2) Discuss the increased stability with stronger overlap integral.
All of the movies listed are accessible by the campus Teaching Resources server. The movies should be shown in class, with the professor offering a play-by-play commentary or step-by-step analysis. Because of the computational capabilities of many of the computes on campus, the suggested viewing arena is Half Sized, and frame by frame motion.
The following images are portions of the saved files for all twelve movies generated for the primer. All of the movies are saved on the Teaching Resources Server under Collaborative Projects/ Chemistry/ Faculty/ CH241/ CH241-movies.
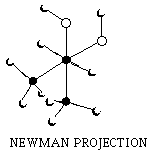
Butane.movie.good copy
newman.2
cyclohexane4596
adamantanemovie
sn2.ch3-cl-br.movie
sn2.-br-oh
sn2.movieCl-F
cp Dimerization
cp Dimer cracking
waterA2virb
waterA1virb
water.B1virb
APPENDICES Appendix A - Acknowledgments
The great increase in computational chemistry used in the classroom of the world has been due to thousands of unsung professors, research groups, and students. This primer is dedicated to everyone concerned with improving education and further increasing the level of scientific knowledge.
Middlebury College has provided me with opportunity to contribute to the curriculum of organic chemistry. For this, I thank the school and all involved. I would like to thank, more specifically, the following people for their support and enthusiasm for education:
Janet Nelson for aiding my exploration of both molecular modeling and education as my future field of work
Jeffrey Byers for the interest in molecular modeling not only at the level of/242, but in research as well
Steven Sontum for understanding the computational and mathematical aspects of molecular modeling as well as supporting my interest in providing this primer to organic chemistry
The seniors class of 1996 and the Febs of 1996.5 for all the entertainment
Anyone who adds to this educational tool, including the organic chemistry students, past and present.
My parents, my sister and Christina, for all their support, encouragement and for understanding that chemistry is something that everyone can learn.
Appendix B - Mouse Key
SGI MacX
Left button ---> mouse button
Middle button -> only left arrow
Right button --> only right arrow
NOTE- When using this table, push the keyboard button then the mouse button. (Ex. Shift then right mouse (arrow) to zoom)
Keyboard
nothing
picking
x/y rotate
x/y translate
space bar
space and control
spc, cntrl, shift
---
---
---
rotate bond
global rotate
z rotate
---
global translate
z translate
Appendix C - Movies
Left
Button
Middle
Button
Right
Button
shift
---
z
rotate
zoom
all
Appendix D - Making Movies
The movies were made by a six step process which was first designed by Matthew Sontum. The process was as follows: make the molecule and all the necessary configurations of the molecule for a movie with SPARTAN, use the Snapshot desktop accessory to save the positions of the molecules as .rgb files, make the movie by placing successive picture after each other with the program MovieMaker, "Fetching" the movie onto the a Power Macintosh 7600 through the Ethernet, and finally viewing the assembled movie on the Macintosh computers.
To transfer all of the movies from the Silicon Graphics Workstations to the Macintosh computers and the Ethernet, the following procedure was followed:
(1) Select "Movie Export Options" under the File menu sub-menu "Export". (2) Choose Apple Quick Time and Apple Animation [RLE] as Compression (3) Select "Export > Movie File..." The movie was then taken from the Silicon Graphics to the Macintosh computers by the Macintosh program Fetch. Once Fetch was opened the following directions were followed:
(1) Enter into the text field that first appears in Fetch:
Host: Alta1
User ID: (e.g. stewart)
Password: my password
Directory: left blank
File Type : Binary
(2) Find the desired movie file from the unix directory. (3) Click the Get button. This will bring up a text field where specific Type and Creator can be established. For all the movies transferred the Type was "MooV" and Creator was "TVOD". The movie was then saved into the Ethernet by placing it into the file Teaching Resources/CollaborativeProjects/Chemistry/ Faculty/ CH241/ CH241-Movies.
Appendix E - Molecular Mechanics, Semiempirical, Ab Initio
Molecular Mechanics:
The parameters for each of the energy terms in a molecular mechanics calculation may vary between individual front end packages, but Equation 1 is the basic form for minimization and molecular mechanics calculations. Other terms, abbreviated "etc." are allowable. As an example, Eelectrostatic is occasionally used to include the electrostatic potential of each atom within the molecule.
The equations for determining the individual energy values are also in standard form throughout several programs. In most modeling programs Molecular Mechanics calculations use Equation 1 to determine the total energy of the molecule.[*] Note that other parameters can be input to influence or justify other chemical traits.
Etotal = Estretch + Ebend + Etorsional + Eout-of-plain + Evan der Waals Eq.1
Estretch is the energy needed to stretch or compress a bond.
Chemically, this parameter is very high. The force constant,
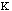 ,
in the Equation 2:
,
in the Equation 2:
Estretch =
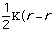 o)2
Eq.2
o)2
Eq.2
is very high. The variable
 is the bond length and
is the bond length and
 o
is the equilibrium value for the molecule.
o
is the equilibrium value for the molecule.
The energy to bend a bond, Ebend , is not as stringent as the
stretching constraints, but allows for minor movement in the molecule. The
force constant,
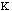 ,
in the Equation 3:
,
in the Equation 3:
Ebond =
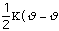 o)2
Eq.3
o)2
Eq.3
is moderately ranged. The angle between the atoms in a molecule is represented
as
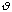 .
The equilibrium angle is represented by
.
The equilibrium angle is represented by
 o.
o.
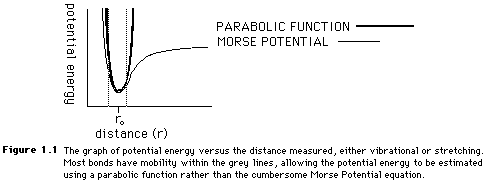
Both the stretching and bending values can be estimated with a parabolic function because of the similarities between the Morse Potential plot of potential energy and the parabolic functions. Figure 1.1 shows the overlap of the two curves. In Figure 1.1, the dotted lines show the average range of bonding stretches. Because the mobility of many molecules is limited, the parabola is a fairly accurate means of obtaining a Morse Potential energy value.
The energy for torsional angle adjustment is calculated from: Etorsional
=
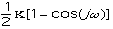 .
The force constant,
.
The force constant,
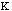 , is very low allowing for free rotation of bonds. This is due to the freedom
that compounds experience in rotations. The variable J has the value of 2 or 3
depending upon the molecules symmetry. The torsional angle,
, is very low allowing for free rotation of bonds. This is due to the freedom
that compounds experience in rotations. The variable J has the value of 2 or 3
depending upon the molecules symmetry. The torsional angle,
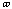 ,
is allowed to change for the calculations.
,
is allowed to change for the calculations.
Out-of-plane vibrational energy, Eout-of-plain, is determined by
the equation: Eout-of-plane =
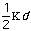 2.
The constant
2.
The constant
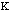 varies from system to system. In molecules with pi bond the constant is high.
The distance from the plane to the molecule of interest is represented by the
variable d.
varies from system to system. In molecules with pi bond the constant is high.
The distance from the plane to the molecule of interest is represented by the
variable d.
The final parameter to be discussed here for molecular mechanics approach to
molecular energy is the van der Waals energy term, Evan der Waals.
This term evaluates the attractive or repulsive forces between atoms. The
equation Evan der Waals =
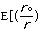 12
12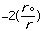 6]
uses the equilibrium distance, ro, the calculated
distance between atoms, r, along with the van der Waals potential of the
interacting atoms,
6]
uses the equilibrium distance, ro, the calculated
distance between atoms, r, along with the van der Waals potential of the
interacting atoms,
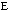 ,
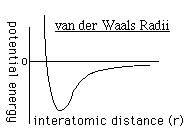
to determine the repulsion or attraction between atoms. Displayed in Figure
1.2 is the relationship described here, a graph of potential energy versus
inter-atomic distances, more commonly known as a van der Waals Radii chart..
The accepted van der Waals radii occurs at the minimum of the curve. Each atom
has its own plot. This brings controversy into the picture because researchers
are unable assign exact measurements for bound versus unbound atoms.
,
to determine the repulsion or attraction between atoms. Displayed in Figure
1.2 is the relationship described here, a graph of potential energy versus
inter-atomic distances, more commonly known as a van der Waals Radii chart..
The accepted van der Waals radii occurs at the minimum of the curve. Each atom
has its own plot. This brings controversy into the picture because researchers
are unable assign exact measurements for bound versus unbound atoms.
FIGURE 1.2
Semiempirical
The above description of a calculation was for molecular mechanics. The force constants, bond lengths, angles and dihedrals are all based upon experimental data. X-ray crystallography provides much of the information for these parameters. As a computational chemist needs grow, for example, they need to know more about a molecule than some distances and angles, the necessary level of theory and computation increases. Semiempirical calculations, as the name implies, is based upon some empirical force field calculations based upon the electrons and molecular orbitals and some parameterized data sets.
The Semiempirical molecular orbital methods are innovative mixtures of experimental data and theoretical expectations. These sets of parameters are generally based upon different families of molecules. One theory may work well with one type of molecule, but very poorly with others. Dependent upon what type of molecule is under investigation, the correct Semiempirical method can be determined from available tables.(6)
The parameters for each method are determined from experimental data. This data allows the program to estimate certain difficult integrals from the Schrödinger time-independent equation. These estimations give results that follow the experimental rather than the theory. They also greatly decrease the computer time.
The introduction of the Neglect of Differential Overlap (NDO) Theory of the 1960's brought Semiempirical theory to molecular modeling programs. From the original set of parameters, several other methods have developed. The two discussed below, Austin Method 1 (AM1) and Parametric Method 3 (PM3), are alternately used for calculations throughout the tutorial.
A discussion of a preliminary Semiempirical method, Modified Neglect of Diatomic Overlap (MNDO), is necessary to comprehend the expansion that the AM1 and PM3 methods offer. Table 1 shows that AM1 and PM3 offer more consistent results than MNDO. The paper published by M. J. Dewar and W. Theil in 1977 offer this objective: "The primary objective of the work reported in this series of papers has been the development of a quantitative treatment of molecular properties accurate enough, reliable enough, and sharp enough to be of practical value in chemistry, in particular in areas where experimental data are lacking or where current experimental procedures fail."(7) The goal was to provide more complete range for calculations and to improve what was currently available.
When the paper was written, there were several quantum mechanical problems with the current methods. Approximations neglected electron repulsion integrals involving a one-center overlap, thus providing rather erratic results. Earlier methods combined twenty two bicentric integrals into one estimated integral. Still other methods assumed that relationships between one centered integrals hold throughout the molecule.
The MNDO Theory, therefore, included four key improvements in the aspects of molecular orbital discussion and calculations. The total energy Etotmol of the molecule is the sum of the electronic energy Eel and the repulsions EABcore between the atoms A and B.(7)
The mathematics involved in calculating the EABcore value are estimated from experimental results. This fact allow researchers the freedom to manipulate the parameter set to fit their experimental designs. If a trend is suspected in a family of molecules, experimental data can be input for the calculations and the math, being based upon experimental results, will show conclusive results. Those molecules that fit the proposed pattern give reasonable results, while those that might appear in the same category, but are actually in a different class of molecules, show dramatically different results. Certain small deviations in bonding energies, bond lengths or heats of formation occur because of the experimental results fusing with theoretical expectations.
The elements H, C, N, O were parameterized by 1985.(8) This broadened the scope of calculations to several organic molecules. The parameters were determined by a nonlinear least-squares optimization process based upon an algorithm devised by Bartels.(9) From this point the mathematics and computations become excessive. The methods were built upon the growing experimental data as well as the theoretical growth of the quantum mechanics. MNDO Theory rests as a primary set of parameters for molecular orbital methods of calculation.
With the understanding that calculations are based on experimental data and mathematical theory, AM1 and PM3 offer parameteric improvements. One of the larger problems of the MNDO theory was its neglect of hydrogen bonding. For better results, a new term had to be added. Simply changing the parameters would not suffice because the MNDO theory had not accounted for such interactions in any way. AM1 was created to be a stepping stone in producing the ideal method.
The Parametric Method Number 3, (PM3) was generated by using an automatic optimization routine. J. J. P. Stewart, in 1989, published "Optimization of Parameters for Methods."(9) This new method of determining parameters involved the first- and second-derivatives of all calculated values for reference data. This step greatly decreased the computational time, thus allowing more data to be handled in the same amount of time.
AM1 is widely used in the primer because of its ability to generate reliable geometries, charge distributions, spin distributions, heat of formations, dipole moments and hydrogen bonding. As of 1990, AM1 is parameterized for 12 elements. Because of this void in complete coverage of all elements further theory may be need by the user. PM3(tm) is used to complete some of the calculations on organometallic complexes. As of SPARTAN version 4.0, AM1 is parameterized for 17 elements: H, B-F, Si-Cl, Zn, Ge, Br, Sn, I. Because of this void in complete coverage of all elements further theory may be need by the user. PM3(tm) is used to complete some of the calculations on organometallic complexes which have Ti, Zr, Hf, Ta, Cr, Mo, W, CO, Rh, Ni, or Pd in them.
Ab Initio
The third possible theory in calculation is the Ab Initio or first principles theory. The calculational programs now have the capacity to accurately estimate the integrals involved in the time-independent Schrödinger Equation:
 =(-h2
=(-h2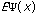 /
2m) (d2
/
2m) (d2
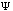 (x)/dx2)
+ U(x)
(x)/dx2)
+ U(x)
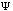 (x).
Eq.4
(x).
Eq.4
These calculations now deal with electrons and their position relative to their velocity. The basic premise behind Ab Initio calculations is this: it uniformly applies a continuous and unbiased field to molecular systems giving energies and kinetics of reactions.(10) Because these methods are not involved in any part of the primer, suggested readings for Ab Initio calculations are included as reference.(11)
Calculations can therefore be placed into three categories. Molecular Modeling, Semiempirical, or Ab Initio. It is imperative that the student understand the results that the computer generates. How the answers were generated is of importance as well, but the final answer must be understood to gain knowledge from the exercises.
Appendix F - SPARTAN Tutorial for Middlebury Campus
Introduction to SPARTAN 4.0
Use of the Three button Mouse and the Mac Mouse:
If you are at an SGI machine there will be a three button mouse.
If you are at a Mac, you will have to use the arrow keys to convert
the one button mouse to a three button one.
SGI Mac
Left button ---> the mouse button
Right button --> the right arrow key
Middle button -> the left arrow key
Keyboard
---
picking
x/y rotate
x/y translate
space bar
space & control
spc, cntrl, shift
---
---
---
rotate bond
global rotate
z rotate
---
global translate
z translate
Acclimate
yourself to moving the molecule with the mouse. Refer to the chart for a one
button mouse and a three button system when necessary. View the molecule down
various bonds and through planes of symmetry.
Left
Button
Middle
Button
Right
Button
shift
---
z
rotate
zoom
all
Visualizations of Molecules:
Once a molecule is on the main screen open the Model menu and look at the different styles of molecular depictions. Select the type of view by clicking it with the left mouse button. The space filling model is of particular interest because it closely resembles the electron density of the molecule. (Note the graphics of the Macs has much lower resolution and poorer quality than the Silicon Graphics Indy Workstation). Using the space filling representation continuously, however, slows the computer down; it is easiest to manipulate the molecules in either the ball and stick or tube models. Again, look down some bonds to get used to the manipulation of the molecules.
How to Build Molecules:
Select New from the File menu. This will bring you into the Builder. There will be two screens present. The one on the left is the work space and is blank. The smaller one, on the right, has some hybridized atoms, a text box and some commands.
To build any molecule you must select the atoms by clicking, with the left mouse button, first on their symbol, then in the work space. Each yellow line seen on the atom represents an open bonding valence. To make a bond click on the atom to add and then on any yellow line with the same hybridization. For example, an sp3 C with an sp3 O, or an sp2 N with an sp2 C Any unfilled yellow lines will be replaced with hydrogens when the molecule is saved so there is no need to add them.
After building the molecule, click the Minimize button in the command box (lower left corner of right hand box.) To Save a built molecule, select Save As from the File menu. Save the molecule by clicking in the text box and type "name of the molecule.yourinitials.todays date"(be sure not to leave spaces) Click save. To quit the builder, select quit from the File menu. The molecule will appear on the original SPARTAN screen.
Building with Groups and Rings:
Enter the builder by opening the File menu and selecting New. To build cyclohexane click on the box marked rings and the word phenyl will appear to the right with a picture of a phenyl group. Click on the button next to the word Phenyl and select the ring you want. The same directions are followed to select a group except that the group button is depressed on the screen.
Building with the Expert Builder:
To enter the Expert Builder select New from the File menu and click Expert from the command box that appears. A new selection of atoms appears and many more valences are possible. Keep in mind the hybridization of each atom when you build in the expert builder. Building is the same in Expert as it is in Entry level building. Again, be sure to minimize each molecule. Select Quit from the file menu and click Yes when it asks if you want to save the molecule.
Making Distance Measurements
Once a molecule is on the main screen open up the Geometry menu and select Distance. Measure the distances around the molecule by clicking on either the bond between two atoms or on two separate atoms. The selected atoms will become yellow and the distance will appear in the upper left hand corner of the screen. You can measure the distance any two atoms, bonded or not. The measurements of molecules is only helpful if you run a calculation on the molecule. Minimizing the molecule is not sufficient for determining such lengths.
Making Angle Measurements
Once a molecule is on the main screen open up Geometry menu and select Angle. To measure an angle, select three adjacent atoms by clicking once with the left mouse button. To unselect an atom click on it again. A selected atom will become yellow. A data box will appear giving you the angle measurement in degrees. The measurements of molecules is only helpful if you run a calculation on the molecule. Minimizing the molecule is not sufficient for determining such angles.
Making Dihedral Angle Measurements
Once a molecule is on the main screen open up Geometry menu and select Dihedral. To measure any dihedral angle (the angle of rotation along a bond), select four adjacent atoms by clicking once with the left mouse button. To unselect an atom click on it again. A selected atom will become yellow. A data box will appear giving you the angle measurement in degrees. The measurements of molecules is only helpful if you run a calculation on the molecule. Minimizing the molecule is not sufficient for determining such angles.
NOTE: When taking any measurement the order in which the atoms are selected is important.
Setting Up a Calculation
Once a molecule is on the main screen open the Model menu and look at the different styles of molecular depiction. Select the type of view by clicking it with the left mouse button. The space filling model is of particular interest because it closely resembles the electron density of the molecule. Using this representation continuously, however, slows the computer down. It is easiest to manipulate the molecules in either the ball and stick or tube model.
Select the molecule to calculate by clicking with the left mouse button once it is on the main screen. The title of the molecule will appear at the top of the SPARTAN screen. Open the Setup menu and select calculational theory you wish to use. This is usually Semiempirical or Molecular Mechanics, DO NOT run Ab Initio calculations unless instructed to by the professor as they take up useful computer time.
A new text box will appear and you must now specify what type of calculation you wish to perform. If you would like to title the calculation, click with the left mouse button, inside the box to the right of the Title: prompt and type your title. It is not necessary to title each calculation. At the Task prompt click on the box and select the appropriate task, quite often it is Geometry Optimization. At the Level prompt click on the box and select the appropriate level of calculation, quite often this is AM1. Enter any options or solvent effects as instructed. The Charge and Multiplicity should also be entered (usually 0 and 1 respectively). With the left mouse button click Save after the correct calculation perimeters have been set up. This will return you to the main SPARTAN screen. You can now setup any Properties, Surfaces, or Volumes by selecting the proper choice under the Setup menu.
To submit the calculation and conformation search open the Setup menu again and select Submit. This will tell the computer to start calculating. Click OK when the text box "Your job 'molecule X.' is running" In a moment the calculation will be done and you will be notified. Click OK when the text box "Your job 'molecule X.' has completed."
Closing SPARTAN:
Open the File menu and select Quit. A text box will appear asking to close all molecules, if any were open, click yes. Quit MacX also. Appendix G - References
1 A SPARTAN Tutorial, Ed. Hehre, W. J.; Burke, L. D.; Shusterman, A. J. Wavefunction, Irvine CA, 1993, p 35.
2 New York Times, Dec. 30, 1995, v145, p23(N), p43(L).
3 Williamson, K. L. Macroscale and Microscale Organic Chemistry Experiments, D. C. Heath, Lexington, MA, 1994, Chapter 12.2.
4 I. Flaming, Selected Organic Syntheses, (John Wiley & Sons, NY, 1977), p 150.
5 Williamson, K. L. Macroscale and Microscale Organic Chemistry Experiments, (D. C. Heath, MA, 1994), Chapter 27.
6 Reviews in Computational Chemistry, Ed. K. B. Lipkowitz; D. B. Boyd, Vol. I, (VCH, New York, 1990), p63.
7 Dewar, J. S.; Theil, W. J . Am. Chem. Soc. 1977, 99, 4899.
8 Bartels, R. H.; University of Texas Center for Numerical Analysis, Report CNA-44, Austin, Texas, 1972
9 Stewart, J. J. J . Comput. Chem., 1989, 10, 209.
10 Clark, T. A Handbook of Computational Chemistry; (John Wiley and Sons, New York, 1985).
11 Hehre, W. J.; Radom, L.; Schleyer, P. v. R.; Pople, J. A. Ab Initio Molecular Orbital Theory (John Wiley & Sons, New York, 1986.)
Appendix H - Suggested Reading
- Reviews in Computational Chemistry, Ed. K. B. Lipkowitz: D. B. Boyd, Vols. I, II, III ,(VCH, 1990). - A SPARTAN Tutorial, Wavefunction, Inc., Irvine, CA, 1993.
- User's Guide, SPARTAN Version 4.0, Wavefunction, Inc., Irvine, CA, 1995.
- Clark, T. A Handbook of Computational Chemistry; (John Wiley and Sons, New York, 1985).
- Hehre, W. J.; Radom, L.; Schleyer, P. v. R.; Pople, J. A. Ab Initio Molecular Orbital Theory (John Wiley & Sons, New York, 1986) - Pople, J.; Beveridge, D. Approximate Molecular Orbital Theory, (McGraw-Hill, NY, 1970)
- Turner A. Methods in Molecular Orbital Theory, (Prentice-Hall, NJ, 1974) - Williamson, K. L. Macroscale and Microscale Organic
Experiments, 2nd Ed.,(D. C. Heath & Co., MA), chapter 16.
Appendix I - Log-in/out Procedure for Spartan (SGI)
To Log In:
1) A green light should be glowing in the upper left hand corner of the blue metallic case. If the light is red, push the round button next to the light. AND record that it was turned off in the log book.
Never turn the computers in 405b off you may unknowingly kill jobs running in the background of the computer.
2) If the monitor is blank and an orange light is showing in the lower right hand corner of the monitor, move the mouse to deactivate the screen saver. In a few moments the screen will come up and a list of files should appear. (If there is a small box labeled "Toolbox" showing, the computer is already logged in. Skip to step four.)
3) Click the on the folder labeled "Orgo", hit return and enter the password given out by your professor.
4) The screen should have a blue background and the mouse will show as an "X". Once the screen with a Toolbox text box appears select "Unix Shell" under the Desktop menu.
5) A blue screen with a prompt "alta#%" will appear on the screen. Place the mouse within the blue screen and do the following:
1) type "rlogin mml#@alta1" (# = integer from 2-13, be sure to note which number you select for future reference)
2) Strike the return key and a password prompt will appear. 3) Enter the password and hit return.
6) The computer has now been instructed to login to alta1 and it will automatically boot up SPARTAN once you have submitted the password.
To log out:
1) Choose Quit from the File menu of SPARTAN.
2) Select Log Out from the Desktop menu in the "Tool box"
3) Click YES at the question: "Do you want to log out now?"
Appendix J - Answer Key:
Teachers Edition:
Student work is shown where the students did the actual assignment.
Date: 05 / 20 /96
Organic Chemistry I
Molecule
|
Carbon
Hybridization
|
C-C
Bond Length (Å)
|
| ethane
.............................
|
___sp3__________
|
___1.5312______
|
| ethene
.............................
|
___sp2__________
|
___1.3373______
|
| ethyne
.............................
|
___sp___________
|
___1.2108______
|
Draw ethane here:
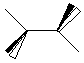
Draw ethene here:
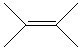
Draw ethyne here:
Chapter 2: Hydrocarbons (#2) Name: ___KEY_______
Date: 05 / 20 /96 Organic Chemistry I
Chapter 2: Hydrocarbons (#3) Name: ___KEY_______
Date: 05 / 20 /96 Organic Chemistry
DRAW & REPORT BOND ANGLES BELOW:
Draw cyclohexane and label the bond angles here:
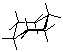 C-C-C bond deg. = 111.8563deg. H-C-H bond deg. = 107.5427deg.
C-C-C bond deg. = 111.8563deg. H-C-H bond deg. = 107.5427deg.
Draw hexane and label the bond angles here:
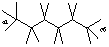
H-C1-H, H-C6-H =107.87deg. carbon 1-2-3, 4-5-6= 111.83deg.
H-C2-H, H-C5-H =107.29deg. 2-3-4, 3-4-5= 111.99deg.
H-C3-H, H-C4-H =107.36deg.
Draw cyclopentane and label the bond angles here:
H-C1-H=108.54deg. carbon 1-2-3, 4-5-1= 103.8deg.
H-C2-H, H-C5-H =107.98deg. carbon 2-3-4, 3-4-5=
H-c3-H, H-C4-H =107.19deg. carbon 5-1-2 = 102.206deg.
Draw pentane and label the bond angles here:
carbon 1-2-3, 4-5-6 = 111.73deg. H-C1-H, H-C5-H = 107.87deg.
2-3-4, 3-4-5 = 111.85deg. H-C2-H, H-C4-H = 107.30deg.
H-C3-H = 107.99deg.
Draw cyclobutane and label the bond angles here:
H-C-H = 112.46deg. C-C-C = 88.08deg.
Draw butane and label the bond angles here:
C-C-C = 111.85deg. H-C1-H = 112.46deg.
TABLE 1
Molecule
|
Heat
of Formation (kcal/mol)
|
| cyclohexane
........................
hexane ...................................
|
__-31.028______
__-44.821_____
|
cyclopentane ......................
pentane .................................
|
__-28.796_____
__-37.473_____
|
cyclobutane ........................
butane ...................................
|
__-0.9868_____
__-31.126_____
|
Date: 05 / 10 /96 Organic Chemistry
Date: 05 / 20 /96 Organic Chemistry I
Molecule: none water
1-propanol, -70.890 -74.749
2-propanol, -71.654 -73.578
1-butanol, -76.320 -79.494
2-butanol, -74.203 -77.587
1-pentanol, -83.171 -86.168
2-pentanol, -82.743 -86.437
3-pentanol. -80.729 -84.010
Chapter 3: Hydrogen Bonding Name:_KEY_______
Date: 05 / 20 /96 Organic Chemistry I
The Heat of Formation is: -119.770603 kcal/mol
The dihedral angle (O-C-C-O) measures to -62.9506deg..
CHAPTER 4- Dipole Moments
Name: ___KEY_______
Date: 05 / 20 /96 Organic Chemistry I
Possible molecules:
dichloromethane, 1.503264 D
trichloromethane, 1.156922 D
tert-butyl chloride, 1.902525 D
dimethylsulfoxide (DMSO), 1.801444 D
acetone, 2.922037 D
benzene. 0.000 D
Why are solvents like DMSO and acetone are used? What intermolecular factors must be taken into account when dealing with these solvents?
Solvents are used because of their polarity or their ability to protonate. Four categories of solvent stem from this generalization: Polar aprotic, polar protic, non-polar aprotic and non-polar protic solvents.
The forces involved with the polarity of the solvent and the solute are van der Waals forces, dipole-induced-dipole interactions and the London force. Van der Waals forces are attractive forces between neutral molecules. Dipole-induced-dipole interactions deals with the small electric field surrounding a molecule with a dipole moment. When this field interacts with a second molecule, the second molecule will have an induced dipole moment. London forces is the dispersion force in an atom when the electronic distribution in two atoms alter slightly.
CHAPTER 4: Dipole Moments Name: ___KEY_______
Date: 05 / 20 /96 Organic Chemistry I
C O Dipole moment = 0.061221 D
What provides this molecule with the decrease in dipole moment?
Resonance structures.
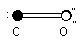
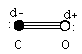
What effects does this decrease have on carbon monoxide's stability and reactivity?
The resonance structures add stability to the molecule. It will allow different localizations of charge. The electrons surrounding the molecule have the ability to group around either the carbon or the oxygen. The oxygen is favored because of its higher electronegativity.
CHAPTER 6- Elimination Reactions Name: __KEY___
Date: 05 / 20 /96 Organic Chemistry I
Find the heat of formation of each alkene in the literature. If the values are different than those you calculated, which are more accurate? The literature is more accurate because the values are based upon the actual chemistry of the molecules. SPARTAN uses molecular theory to determine the heats of formation.
Can we still use the values SPARTAN calculated? Yes, what SPARTAN does offer is a relative stability when comparing the three products. The difference in energy will show the relative stabilities.
Date: 05 / 20 /96 Organic Chemistry II
What does this fact imply about resonance stability?
The carbonyl cannot accept electron density from the cyclopentadiene ring.
The [Delta]H of the out of plane conformation is 15.130 kcal/mol.
The [Delta]H of the in plane conformation is 17.505 kcal/mol.
Date: 05/20/96 (3/18/96) Organic Chemistry II
cyclohexadienone:
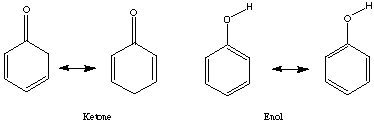
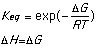
cyclohexanone: cyclohexandione:
ketone: [Delta]H = -63.33405 kcal/mol ketone: [Delta]H = -63.33405 kcal/mol
enol: [Delta]H = -55.897371 kcal/mol enol: [Delta]H = -19.809117 kcal/mol
[Delta]H:
enol -ketone = enol -ketone =
+7.43668 kcal/mol -11.174780 kcal/mol
= 31,115.069 J/mol = -46,755.28 J/mol
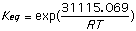
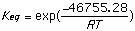
= 3.5142x 10-6 = 1.57x108
Using Spartan and the shown calculations we produced Keq values consistent with trends demonstrated by the literature values. The discrepancy probably arises because different values for [Delta]H were obtained experimentally and used to calculate Keq than those obtained by Spartan. Spartan neglects to account for the 2,5 cyclohexadienone, a major resonance contributor which when accounted for could explain the higher Keq cited in the provided literature values. The Keq for cyclohexanone was very close to literature because no major resonance structures were excluded by Spartan. The small (<1) Keq for cycloketone suggests that the ketone form is far more stable that the enol form. The higher Keq (>1) for cyclohexandianone indicates stability lies with the enol form. this is caused by the fact that the "ketone -> enol" allows for more stabilizing resonance structures. Essentially, it is a phenyl ring with a hydroxyl substituent (phenol).
CHAPTER 9 - Infrared Spectroscopy
CHAPTER 9
Infrared Assignment Name: _______KEY___
Date: 05 / 20 /96
CH 241
C=O : Frequency (cm-1)
Spartan Literature
formaldehyde, 1987.29 1650
acetone, 2063.16 1710
cyclobutanone, 2164.46 1775
cyclohexanone, 2065.54 1710
2,3-hexanedione.
both up, both down= 2071.93 1710(s)
alternating= 2065.62 1690(m
Compare and contrast:
A: acetone and formaldehyde,
Acetone has a higher frequency than formaldehyde. This runs parallel with the literature values above.
B: cyclohexanone with cyclobutanone,
The added strain energy of cyclobutanone increased the vibrational energy of the carbonyl stretch when compared to the cyclohexanone. The pattern is again confirmed by the literature.
and C: the two frequencies of 2,3-hexanedione.
The both up and both down vibration (|| - ||, '' - '') was slightly higher in energy than the alternating group (|| - '' , '' - ||). This is comparable to the literature values listed above. The added momentum of both carbonyls vibrating in rhythm might add to the energy.
1) Observe the overhead of HOMO and LUMO overlap of acrolein and butadiene.
2) Discuss the increased stability with stronger overlap integral.
The carbonyl on the acrolein has a larger overlap when the LUMO of acrolein is arranged pointing toward the butadiene.
The overlap is less when the carbonyl points away from the diene.
[*] Look in any currnet issue of The Journal of Chemistry in Education or Jouranl of the American Chmical Sociaty.
** For a more complete investigation see Appendix C, or Refercne (1)
[*] Williamson's text offers a silimar discussion in Chapter 16.